Другие заболевания двигательных нейронов у детей - кратко с точки зрения педиатрии
Заболевания двигательных нейронов представляют собой гетерогенную группу прогрессирующих нейродегенеративных расстройств, характеризующихся дисфункцией верхних и нижних нейронов с началом от рождения до зрелого возраста. Следует учитывать разл. причины, включая наследственные, иммуноопосредованные, инфекционные, паранеопластические и спорадические заболевания.
Острый вялый паралич — наиболее распространенное проявление заболеваний двигательных нейронов у детей; он может возникать в виде вспышек. Полиомиелит раньше был основной причиной хронической инвалидности, но при рутинном использовании полиовакцины эта вирусная инфекция встречается редко.
Др. энтеровирусы, такие как вирус Коксаки и эховирус, или живая вакцина против полиомиелита также могут вызывать острую инфекцию двигательных нейронов с симптомами и признаками, похожими на полиомиелит, хотя обычно более мягкими. Диагностическими являются специфические ПЦР-тесты и вирусные культуры СМЖ. Сообщалось о скоплении случаев острого вялого паралича во время вспышек энтеровируса D68 в нескольких состояниях у детей (средний возраст 7-11 лет).
Слабость конечностей часто асимметричная и включает бульбарную слабость, а также поражение VI и VII ЧМН. МРТ может выявлять продольные поражения спинного мозга с доминирующим вовлечением клеток переднего рога (рис. ниже). Плеоцитоз СМЖ и повышенный уровень белка являются общими. Лечение включало стероиды и в/в-Ig; персистирующий парез является распространенным последствием. Происходит также поражение двигательных нейронов вирусом Западного Нила.
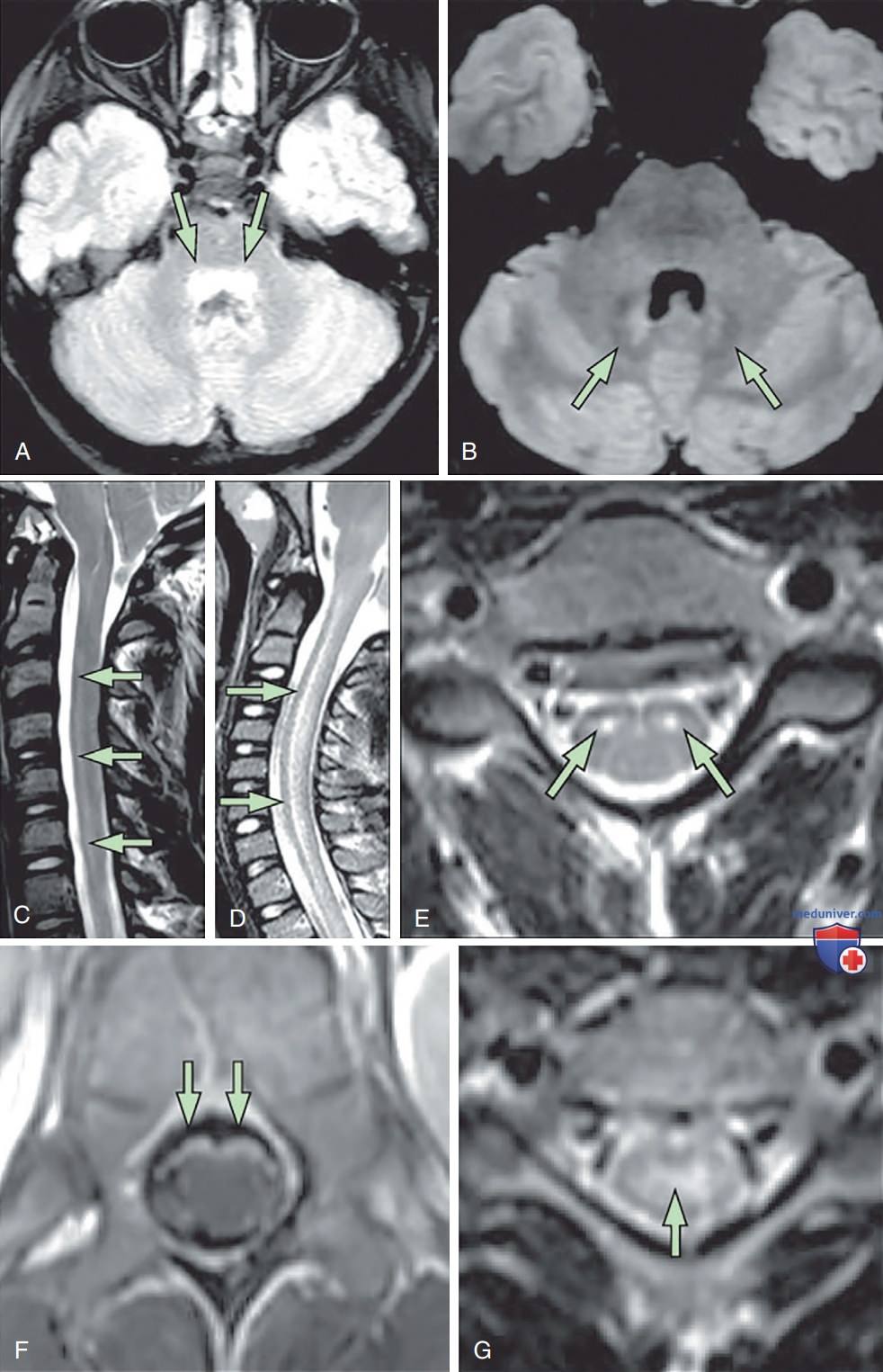
Магнитно-резонансная томография головного мозга и спинного мозга. Аксиальные FLAIR* на уровне Понса показывает повышенный сигнал в тегментуме (A; случай 2). Аксиальные FLAIR на уровне Понса показывает повышенный сигнал в зубчатых ядрах, правое больше левого, а сагиттальное Т2-взвешенное изображение ШОП показывает гиперинтенсивность длинных сегментов в вентральном канатике (B, C; случай 5). Сагиттальное Т2-взвешенное изображение показывает обширное увеличение сигнала в центральном канале и набухание спинного мозга (D) при предъявлении (D-F; случай 1). Через 5 нед после презентации аксиальная Т2-взвешенная визуализация ШОП показывает остаточный повышенный сигнал в передних рогах (E), а аксиальная Т1-взвешенная постконтрастная визуализация показывает усиление вентрального нервного корешка на уровне медуллярного конуса (F). Осевое Т2-взвешенное изображение ШОП, показывающее повышенный сигнал в центральном сером в-ве (G; случай 6). Случаи 2, 5 и 6 были детьми с энтеровирусом D68, выявленным в носоглотке.
У детей внезапное начало, медленное прогрессирование и семейный анамнез могут быть ключами к генетической основе. Хотя наиболее распространенным заболеваний двигательных нейронов у детей является 5Q13-ассоциированная СМА с типичным или преобладающим фенотипом нижних моторных нейронов, существует клинически и генетически гетерогенная группа заболеваний двигательных нейронов, которые дифференцируются с наследственными спастическими параплегиями, наследственными сенсомоторными невропатиями и ювенильными формами бокового амиотрофического склероза.
Менее распространенная группа заболеваний двигательных нейронов, не связанная с SMN1, называется не-5q13-связанная СМА; эта гетерогенная группа может быть связана с Х-хромосомой, АуД или АуР-СМА, дистальной моторной невропатией, сегментарной моторной невропатией или дистальной наследственной двигательной невропатией либо нейронопатией. Дополнительные признаки, такие как глухота; эпилепсия; энцефалопатия; спастичность; нарушение зрения; стволовые, мозжечковые, желудочно-кишечные или ревматологические расстройства, могут указывать на полиорганное поражение.
Эти атипичные фенотипы СМА также можно назвать синдромами СМА-плюс, и они имеют разные фенотипические признаки и молекулярно-генетическую гетерогенность (табл. 11). Первичное поражение верхнего двигательного нейрона с прогрессирующей потерей верхнего и нижнего двигательных нейронов характеризует ювенильный боковой амиотрофический склероз, который встречается редко и в конечном счете приводит к летальному исходу.
Параллельно с достижениями в области методов следующего поколения наблюдается увеличение молекулярного диагностического выхода и расширение клинических фенотипов. Это дополнительно помогает не только понять естественное течение и общие патофизиол. механизмы, но и указывает на необходимость генетического консультирования и пренатальной диагностики.
Следует оценить характер слабости, амиотрофии и прогрессирования (проксимальное или дистальное, бульбарное или респираторное поражение), наличие спастичности, глубоких сухожильных рефлексов и семейный анамнез. В отличие от типичной СМА, электрофизиол. исследования и ЭМГ могут служить важными инструментами для доказательства нейрогенной основы. Требуется многосистемная оценка, в т.ч. зрения, слуха и когнитивного развития. Клиническая оценка и распознавание отличительных признаков помогут классифицировать заболевания двигательных нейронов и учитывать излечимые формы заболевания двигательных нейронов после ДД.
СМА с РДС — редкое АуР-заболевание, обусловленное мутациями в гене, кодирующем IGHMBP2 на хромосоме 11q13. В отличие от классического СМА I типа, преобладающая дистальная слабость при диафрагмальном параличе приводит к тяжелой ДН. Обычно наблюдается раннее проявление в возрасте от 6 нед до 6 мес с ЗВУР, слабым криком и сосанием, а также врожденными деформациями стопы. Обычная РОГК может выявить диафрагмальную грыжу, которая вызывает раннюю ДН. Описаны также атипичные пациенты с периферической невропатией и отсутствием нарушения дыхания.
Помимо основных симптомов, сенсорная и вегетативная дисфункция (чрезмерное потоотделение, задержка мочи, запор и сердечная аритмия), судороги и прогрессирующее поражение ЧМН могут быть дополнительными признаками.
Синдром Брауна-Виалетто-Ван Лаэра — редкое гетерогенное нейродегенеративное заболевание, характеризующееся поражением VII—XII ЧМН, прогрессирующей слабостью мышц лица, нейросенсорной глухотой, дисфагией, амиотрофией языка, фасцикуляциями, бульбарным параличом и ДН. Он может развиваться в любом возрасте. Слабость рук и кистей, атрофия зрительного нерва и атаксия могут быть дополнительными симптомами. Клиническая картина синдрома Фацио-Лонда одинаковая и характеризуется прогрессирующим бульбарным параличом, возникающим в результате дегенерации двигательных нейронов больше в стволе ГМ, чем в спинном мозге, без нейросенсорной глухоты.
Идентификация мутаций в генах-переносчиках рибофлавина (табл. 11) обеспечила целенаправленную терапевтическую стратегию с пероральным или в/в- введением высоких доз рибофлавина в обычной дозе 10 мг/кг/сут. Клинический эффект в этой группе может варьироваться от быстрого ответа до постепенного улучшения в течение 12 мес, клинической стабилизации или редко отсутствия эффекта. Следует принимать во внимание распознавание аномальных профилей ацилкарнитина, имитирующих множественный дефицит ацил-Коа-дегидрогеназы при синдроме Брауна-Виалетто-Ван Лаэра. Биохимический эффект на лечение также очевиден.
Общий основной фенотип этого заболевания двигательных нейронов, которое поддается лечению, включает прогрессирующую аксональную сенсомоторную нейропатию (проявляющуюся сенсорной атаксией и выраженной слабостью верхних конечностей и аксиальных мышц с отчетливо сохраненной мышечной силой нижних конечностей), потерю слуха, атрофию зрительного нерва и ДН.
Классическая форма понтоцеребеллярной гипоплазии со СМА характеризуется выраженной гипотонией, арефлексией, мышечной слабостью, центральным нарушением зрения, дисфагией, ДН и приобретенной микроцефалией, с проявлением в первые месяцы жизни и смертью в младенчестве. Существует широкий клинический спектр, с тяжелым антенатальным началом, представляющим собой тяжелый конец спектра с артрогрипозом и многоводием.
СМА с прогрессирующей миоклонической эпилепсией (SMAPME) характеризуется устойчивой к лечению прогрессирующей миоклонической эпилепсией в сочетании со слабостью проксимальных мышц, арефлексией, атрофией, прогрессирующей слабостью и дисфагией, за которыми следуют нормальные этапы развития. Легкая слабость мышц лица, фасцикуляция языка, нейросенсорная тугоухость и тремор могут быть дополнительными признаками.
Редкие варианты включают недавно описанный полиартикулярный артрит со СМА, умеренный СМА без судорог, эпилептический миоклонический статус век, отсутствие и атонические судороги в подростковом возрасте.
Летальный артрогрипоз с поражением клеток переднего рога и СМА с врожденным артрогрипозом и переломами являются атипичными формами СМА в спектре акинезии/гипокинезии плода.
Разл. митохондриальные заболевания могут иметь СМА-подобный клинический фенотип. В дополнение к гипотонии, слабости и ДН существует более широкий спектр мультисистемного поражения, такого как инфантильная ГКМП, печеночная недостаточность, спастичность, синдром Ли, энцефалопатия, судороги, дисфункция ствола мозга, глобальная задержка развития, птоз и офтальмоплегия. Лактоацидоз и повышенный уровень креатинкиназы в сыворотке крови могут дополнительно помочь выявить гены, участвующие в белках сборки ЦОГ и синдромах истощения митохондрий.
СМА с преобладанием слабости нижних конечностей характеризуется врожденной или ранней проксимальной мышечной слабостью и атрофией нижних конечностей. Существует также широкий спектр клинических проявлений от антенатального до зрелого периода. Спастичность и когнитивные нарушения могут быть частью клинической картины у некоторых пациентов.
Плечелопаточная СМА — АуД-состояние, определяемое избирательным вовлечением мышц, прогрессирующей дистальной слабостью и атрофией. Паралич гортани, нейросенсорная глухота, низкий рост, сколиоз, легкая дисплазия конечностей и скелета могут сопровождать картину.
Двигательные нейроны вовлекаются в некоторые метаболические заболевания НС, такие как ганглиозидоз (болезнь Тея-Сакса), цероидный липофусциноз (болезнь Баттена) и гликогеноз II (болезнь Помпе), но признаки денервации могут быть незначительными или скрытыми с более заметным вовлечением др. частей ЦНС или мышц. Амиотрофия, связанная с дегенерацией нижних двигательных нейронов, является ведущим признаком некоторых мультисистемных расстройств, таких как инфантильная нейроаксональная дистрофия, ахалазия-аддисонианизм-алакримия (тройной А или синдром Олгроува) и синдром Чедиака-Хигаси.