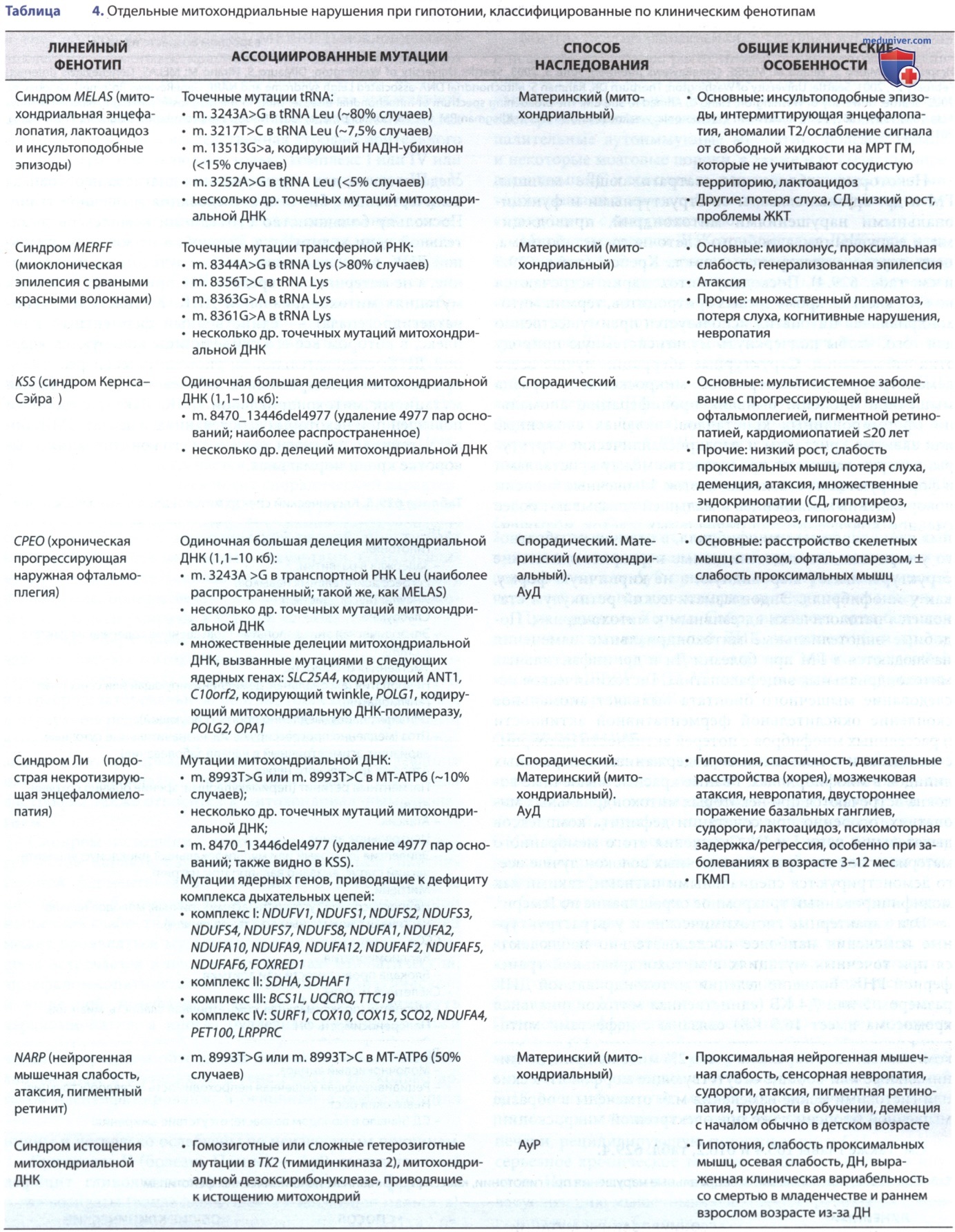
Некоторые заболевания, затрагивающие мышцы, ГМ и др. органы, связаны со структурными и функциональными нарушениями митохондрий, приводящими к нарушениям аэробного клеточного метаболизма, цепи переноса электронов и цикла Кребса (Krebs, Sir Hans Adolf) (табл. 5 и см. табл. 4). Поскольку митохондрии встречаются во всех клетках, кроме зрелых эритроцитов, термин митохондриальная цитопатия используется преимущественно для того, чтобы подчеркнуть мультисистемную природу этих заболеваний.
Структурные аберрации лучше всего демонстрируются электронной микроскопией образца мышечной биопсии, выявляя пролиферацию аномально сформированных кристаллов, включая сложенные или завитые кристаллы и паракристаллические структуры, которые занимают пространство между кристаллами и формируются из стволовых клеток. Мышечные биопсии новорожденных, младенцев и малышей показывают более сильное вовлечение эндотелиальных клеток в/мышечных капилляров, чем миофибрилл, в отличие от обратного у взрослых, но эндотелиальные паракристаллические структуры имеют шаровидную, а не кирпичную форму, как у миофибрилл. Эндоплазматический ретикулум становится патологически адгезивным к митохондриям.
Подобные эндотелиальные митохондриальные изменения наблюдаются в ГМ при болезни Ли и др. инфантильных митохондриальных энцефалопатиях. Гистохимическое исследование мышечного биоптата выявляет аномальное скопление окислительной ферментативной активности и рассеянных миофибров с потерей активности цитохром-с-оксидазы и с повышенным содержанием нейтральных липидов в миофибриллах. Рваные красные мышечные волокна встречаются при некоторых митохондриальных миопатиях, особенно при сочетании дефицита комплексов дыхательной цепи I и IV.
Скопления этого мембранного материала под мембраной мышечных волокон лучше всего демонстрируются специальными пятнами, такими как модифицированный трихромное окрашивание по Гемери (Gomori, Gyorgy).
Эти характерные гистохимические и ультраструктурные изменения наиболее последовательно наблюдаются при точечных мутациях в митохондриальной трансферной РНК. Большие делеции митохондриальной ДНК размером 5 или 7,4 КБ (единственная митохондриальная хромосома имеет 16,5 КБ) связаны с дефектами митохондриальных дыхательных окислительных ферментных комплексов, если поражено всего 2% митохондрий, но минимальные или вообще отсутствующие морфологические или гистохимические изменения м.б. отмечены в образце мышечной биопсии даже при электронной микроскопии; следовательно, для подтверждения диагноза необходимы количественные биохим. исследования мышечной ткани.
Поскольку большинство субъединиц комплексов дыхательной цепи кодируются ядерной, а не митохондриальной ДНК, возможно менделевское аутосомное наследование, а не материнская передача, как при чистых точечных мутациях митохондриальной ДНК. Комплекс II (сукцинатдегидрогеназа) — единственный ферментный комплекс, в котором все его субъединицы кодируются ядерной ДНК; следовательно, он гистохимически реактивен при всех митохондриальных заболеваниях с точечными мутациями митохондриальной ДНК. Лактат сыворотки повышен при некоторых заболеваниях, а лактат СМЖ более устойчиво повышен, даже если его концентрация в сыворотке крови нормальная.
Идентифицировано несколько разл. митохондриальных заболеваний, которые в первую очередь поражают поперечнополосатую мышцу или и мышцы, и ГМ. Их можно разделить на болезни рваных красных волокон и болезни без рваных волокон. К заболеваниям с рваными красными волокнами относятся синдромы Кернса-Сэйра, MELAS (митохондриальная энцефалопатия, лактоацидоз и инсультоподобные эпизоды), MERRF (миоклоническая эпилепсия с рваными красными волокнами) и синдром прогрессирующей внешней офтальмоплегии, которые связаны с сочетанным дефектом комплексов дыхательной цепи I и IV.
К заболеваниям без рваных волокон относятся энцефалопатия Ли и наследственная атрофия зрительного нерва Лебера (Leber, Theodor); они включают только комплекс I или IV или у детей обычное сочетание дефектных комплексов III и V синдром Кернса-Сэйра характеризуется триадой прогрессирующей внешней офтальмоплегии, пигментной дегенерацией сетчатки и началом <20 лет. Часто это сопровождается блокадой сердца, дефицитом мозжечка и высоким содержанием белка в СМЖ. Зрительные вызванные потенциалы изменены. Больные обычно не испытывают слабости туловища или конечностей либо дисфагии. Большинство случаев носят спорадический характер.
Хроническая прогрессирующая наружная офтальмоплегия м.б. изолированной или сопровождаться слабостью мышц конечностей, дисфагией и дизартрией. Отмечено несколько пациентов, описанных как имеющие офтальмоплегию плюс дополнительное поражение ЦНС. АуД-наследование встречается в некоторых родословных, но в большинстве случаев носит спорадический характер.
Синдромы MERRF и MELAS — другие митохондриальные заболевания у детей. Последний характеризуется задержкой роста, эпизодической рвотой, приступами и повторяющимися мозговыми инсультами, вызывающими гемипарез, гемианопсию или даже корковую слепоту и слабоумие. Болезнь ведет себя как дегенеративное заболевание, и дети умирают в течение нескольких лет.
Др. «дегенеративные» заболевания ЦНС также включают миопатию с митохондриальными аномалиями, подострую некротическую энцефалопатию Ли и цереброгепаторенальную болезнь (болезнь Зеллвегера (Zellweger, Hans Ulrich)), в первую очередь пероксисомную болезнь с вторичными изменениями митохондрий. Др. признанной митохондриальной миопатией является дефицит цитохром-с-оксидазы. Окулофарингеальная мышечная дистрофия также относится к митохондриальным миопатиям.
Синдром истощения митохондрий в раннем младенчестве характеризуется резко сниженной окислительной ферментативной активностью в большинстве или во всех 5 комплексах; в дополнение к диффузной мышечной слабости у новорожденных и маленьких детей может проявляться мультисистемное поражение, и синдром встречается в нескольких формах: миопатической; энцефаломиопатической; гепатоэнцефалопатической; и кишечной энцефалопатической. Может возникнуть кардиомиопатия, а иногда буллезные поражения кожи или генерализованный отек.
Синдром Альперса (Alpers, Bernard J.) генетически однороден и обусловлен истощением митохондриальной ДНК и мутациями в гене POLG1. Несколько др. генов идентифицированы, в основном в более поздних формах; следовательно, истощение митохондрий — это синдром, а не отдельное заболевание. Синдром Барта (Barth, Peter) — это Х-сцепленное рецессивное митохондриальное расстройство, характеризующееся кардиомиопатией, миопатией поперечнополосатых мышц, задержкой роста, нейтропенией и высокими концентрациями 3-метил-глутаконовой кислоты в сыворотке крови и моче.
Многие редкие заболевания, о которых известно лишь в нескольких случаях, расцениваются как митохондриальные нарушения. В настоящее время признано, что вторичные митохондриальные дефекты возникают при широком спектре немитохондриальных заболеваний, включая воспалительные аутоиммунные миопатии, болезнь Помпе (Pompe, Joannes Cassianus) и некоторые мозговые пороки, а также м.б. вызваны определенными ЛС и токсинами, поэтому к интерпретации митохондриальных аномалий как первичных дефектов следует подходить с осторожностью.
Митохондриальная ДНК отличается от ДНК ядра клетки и наследуется исключительно от матери; митохондрии присутствуют в цитоплазме яйцеклетки, а не в головке сперматозоида, единственной части, которая входит в яйцеклетку при оплодотворении. Скорость мутации митохондриальной ДНК в 10 раз выше, чем у ядерной ДНК. Митохондриальные дыхательные ферментные комплексы имеют субъединицы, кодируемые либо в митохондриальной ДНК, либо в ядерной ДНК. Комплекс II (сукцинатдегидрогеназа, фермент цикла Кребса) имеет 4 субъединицы, все они кодируются в ядерной ДНК; комплекс III (убихинол или цитохром-оксидаза В) состоит из 9 субъединиц, из которых только один является кодируемым митохондриальной ДНК и 8 из них программируются ядерной ДНК; комплекс iv (цитохром — С-оксидазы) имеет 13 подразделений, только 3 из которых кодируются митохондриальной ДНК.
По этой причине митохондриальные заболевания мышц могут передаваться как АуР-признаки, а не строго материнской передачей, хотя все митохондрии наследуются от матери.
При синдроме Кернса-Сэйра выявлена одна большая делеция митохондриальной ДНК, но известны и др. генетические варианты; при синдромах митохондриальной миопатии MERRF и MELAS точечные мутации встречаются в транспортной РНК.
а) Обследование. Исследование митохондриальных цитопатий начинается с сывороточного лактата. Молочная кислота повышена не во всех митохондриальных цитопатиях, так что нормальный результат не должен обнадеживать; лактат СМЖ повышен в некоторых случаях, когда лактат сыворотки в норме, особенно при наличии клинических признаков энцефалопатии.
Сывороточная 3-метил-глутаконовая кислота часто повышается при митохондриальных цитопатиях в целом, демонстрируется >50 разл. генетических мутаций и, следовательно, является хорошим скрининговым показателем; она редко повышается при др. метаболических заболеваниях. Уровень этого в-ва м.б. увеличен в моче. Печеночные ферменты (трансаминазы) следует измерять в крови. Оправдано обследование ССС. Молекулярные маркеры в крови для распространенных заболеваний с известными точечными мутациями митохондриальной ДНК идентифицируют многие митохондриальные цитопатии, проявляющиеся во взрослой жизни или подростковом возрасте, но реже у детей и у младенцев.
МРТ ГМ может выявить гиперинтенсивные поражения базальных ганглиев, MP-спектроскопия может продемонстрировать повышенный пик лактата. Биопсия мышц дает наилучшие доказательства при всех митохондриальных миопатиях и должна включать гистохимию окислительных ферментов, электронную микроскопию и количественный биохимический анализ ферментных комплексов дыхательной цепи и коэнзима Q10; мышечная ткань м.б. проанализирована на митохондриальной ДНК.
Многие митохондриальные нарушения могут поражать шванновские клетки и аксоны периферических нервов, клинически проявляться невропатией; следовательно, у отдельных пациентов можно измерить скорость проводимости двигательных и сенсорных нервов; биопсия сурального нерва требуется лишь в редких случаях, если преобладает нейропатия и диагноз не подтвержден др. исследованиями.
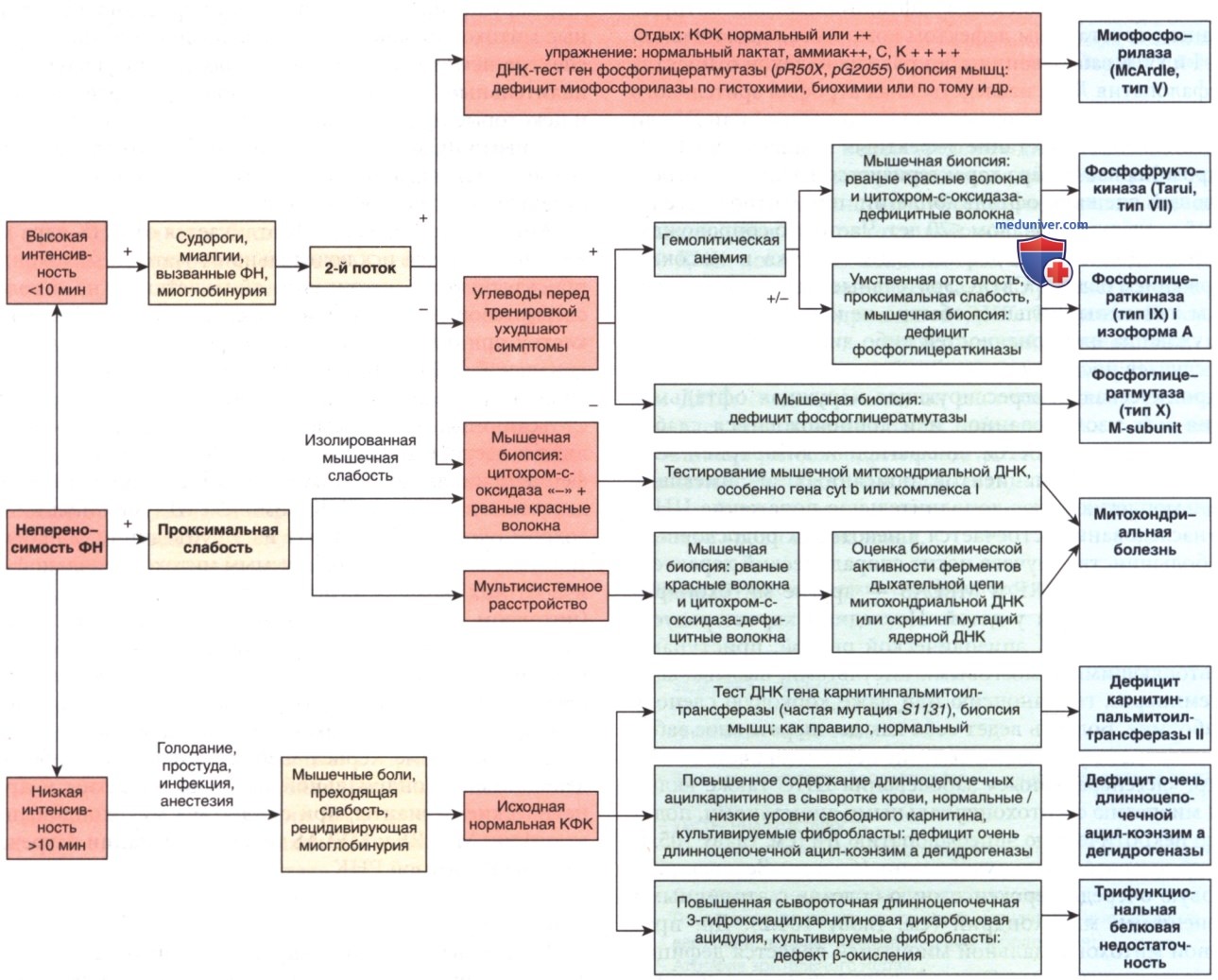
Диагностический подход представлен на рис. ниже.
Алгоритм клинической диагностики пациентов с непереносимостью ФН, у которых подозревается метаболическая миопатия.
б) Лечение. Эффективного лечения митохондриальных цитопатий не существует, но разл. комбинации часто используются эмпирически, чтобы попытаться преодолеть метаболический дефицит. К ним относятся пероральные добавки с карнитином, рибофлавин, кофермент Q10, аскорбиновая кислота (витамин С), витамин Е и др. антиоксиданты. Хотя некоторые спорные сообщения обнадеживают, никаких контролируемых исследований, доказывающих эффективность, опубликовано не было.