а) Аутоиммунная миастения. Миастения гравис — хроническое аутоиммунное заболевание постсинаптической концевой пластинки, приводящее к нарушению нервно-мышечной передачи или блокаде, клинически характеризующееся быстрой утомляемостью поперечнополосатых мышц, особенно экстраокулярных и пальпебральных мышц (мышц века), а также глотательных мышц. Это заболевание следует отличать от врожденного миастенического синдрома, генетического нарушения рецепторов на пресинаптической и постсинаптической мембранах, а также синапса нервно-мышечного соединения, и нарушений нейротрансмиссии вызванных токсинами, таких как ботулизм.
При миастении гравис высвобождение ацетилхолина (АЦХ) в синаптическую щель аксональным концом является нормальным, но постсинаптическая мышечная мембрана (т.е. сарколемма) или моторная концевая пластинка менее чувствительны, чем обычно. Это происходит из-за АТл против постсинаптического рецептора АЦХ, что приводит к аномальной архитекту-ре/складчатости постсинаптической мембраны, а также к уменьшению числа рецепторов, с которыми АЦХ может связываться.
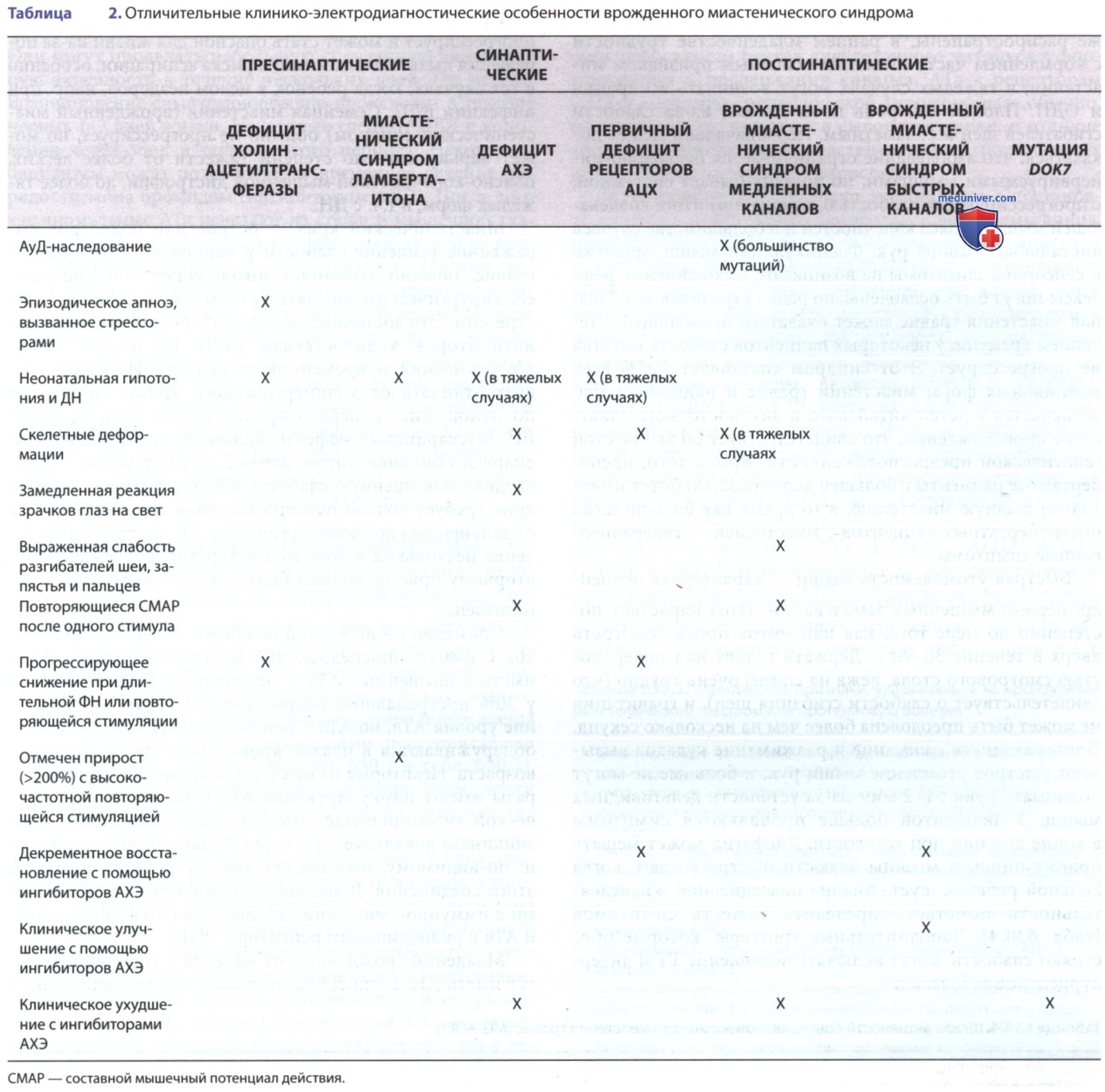
Младенцы, рожденные от матерей с миастенией, могут иметь транзиторный неонатальный миастенический синдром, вторичный по отношению к плацентарно перенесенным АТл к рецепторам АЦХ, отличные от врожденных миастенических синдромов (табл. 1, 2, 3).
б) Клинические проявления. Возраст начала иммуноопосредованной миастении гравис колеблется от 11 мес до 17 лет. В препубертатных возрастных группах М/Ж составляет ок.1,5:1, а в постпубертатных возрастных группах М/Ж — ок. 1:1. При ювенильной аутоиммунной миастении гравис наиболее ранними и постоянными признаками являются односторонний или двусторонний, но обычно асимметричный птоз и некоторая степень экстраокулярной мышечной слабости. Экстраокулярная слабость не ограничивается мышцами, иннервируемыми только одним или двумя из трех соответствующих ядер ствола ГМ; она прогрессирует.
Дети постарше могут жаловаться на диплопию, а маленькие дети могут держать пальцами открытые глаза или только большими пальцами, если птоз достаточно выражен и может затруднить зрение. Зрачковые реакции на свет сохраняются. Дисфагия и лицевая слабость также распространены, в раннем младенчестве трудности с кормлением часто являются основным признаком миастении; в тяжелых случаях могут возникать аспирация и ОДП. Плохой контроль позы головы из-за слабости сгибателей шеи м.б. заметным. Первоначально может показаться, что заболевание ограничивается бульбарно-иннервируемыми мышцами, но болезнь бывает системной, с прогрессирующей слабостью, в конечном итоге вовлекающей мышцы пояса конечностей и в большинстве случаев дистальные мышцы рук.
Фасцикуляции мышц, миалгии и сенсорные симптомы не возникают. Сухожильные рефлексы могут быть ослаблены, но редко утрачиваются. Глазная миастения гравис может оказаться преходящей с течением времени, у некоторых пациентов слабость никогда не прогрессирует. Этот синдром составляет 25% всех ювенильных форм миастении гравис и наиболее часто встречается у детей китайского и юго-восточного азиатского происхождения, что свидетельствует об этнической генетической предрасположенности. Кроме того, препубертатные пациенты с большей вероятностью будут иметь только глазную миастению, в то время как большинство постпубертатных пациентов с миастенией — генерализованные симптомы.
Быстрая утомляемость мышц — характерная особенность миастении гравис, отличающая ее от большинства др. нервно-мышечных заболеваний. Птоз нарастает постепенно по мере того, как пациентов просят смотреть вверх в течение 30-90 с. Держать голову над поверхностью смотрового стола, лежа на спине, очень трудно (что свидетельствует о слабости сгибания шеи), и гравитация не может быть преодолена более чем на несколько секунд. Повторяющиеся сжимание и разжимание кулаков вызывают быстрое утомление мышц рук, и больные не могут поднимать руки >1-2 мин из-за усталости дельтовидных мышц.
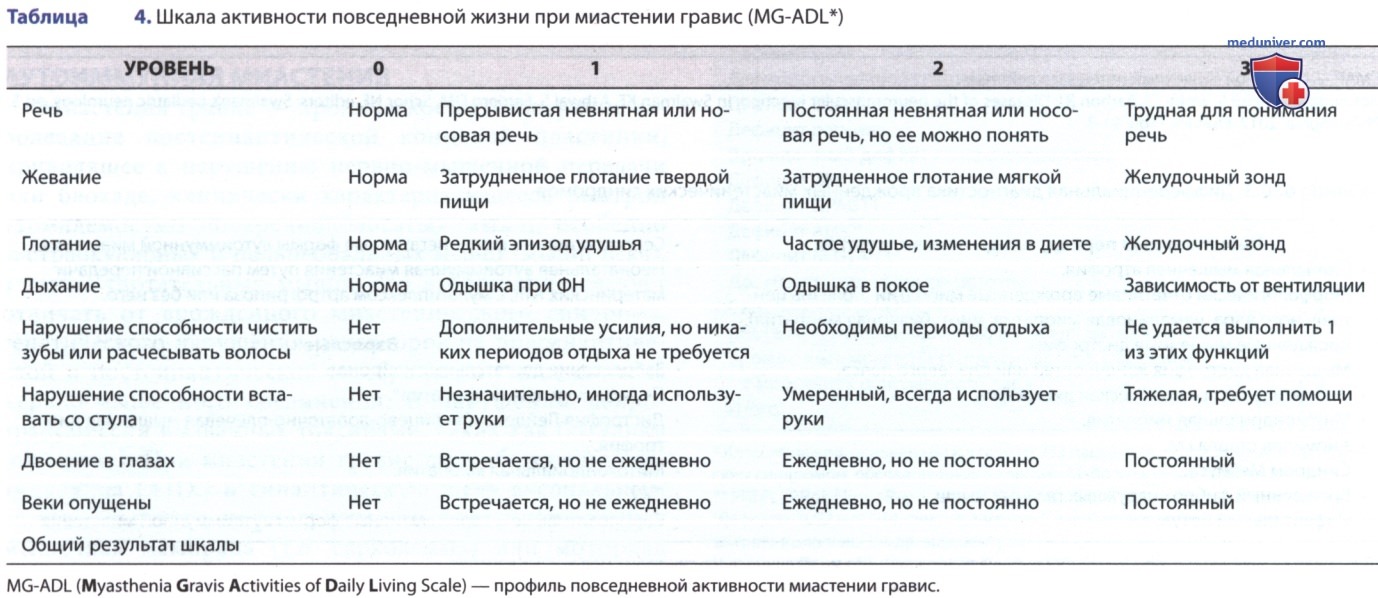
У пациентов больше проявляются симптомы в конце дня или при усталости. Дисфагия может мешать приему пищи, и мышцы челюсти быстро устают, когда больной ребенок жует. Анализ повседневной жизнедеятельности помогает определить тяжесть симптомов (табл. 4). Дополнительные триггеры, которые обостряют слабость, могут включать повышение ТТ и интеркуррентное заболевание.
При отсутствии лечения миастения гравис обычно прогрессирует и может стать опасной для жизни из-за поражения дыхательных мышц и риска аспирации, особенно в тех случаях, когда ребенок в целом нездоров, напр. при инфекции ВДП. Семейная миастения (врожденный миастенический синдром) обычно не прогрессирует, но может варьировать по степени тяжести от более легких, поясно-конечностной мышечной дистрофии, до более тяжелых форм, в т.ч. с ДН.
Миастенический криз — острое или подострое выраженное усиление слабости у пациентов с миастенией гравис, обычно вызванное интеркуррентной инфекцией, хирургическим вмешательством или эмоциональным стрессом. Это состояние может потребовать в/в-введения ингибиторов холинэстеразы (ХЭ), Ig, плазмофереза, в/в-кормления и временной поддержки ИВЛ. Его следует отличать от холинергического криза, вторичного по отношению к передозировке антихолинэстеразным ЛП. Мускариновые эффекты включают спазмы в животе, диарею, обильное потоотделение, слюнотечение, брадикардию, повышенную слабость и миоз. Холинергический криз требует только поддерживающей терапии и отказа от дальнейших доз холинергических ЛП, он проходит в течение нескольких часов; доза лекарства, подлежащая повторному приему, должна быть пересмотрена, если только пациент не принял чрезмерную дозу, которая не была назначена.
Примерно 70-80% детей младшего возраста и подростков с иммуноопосредованной миастенией гравис будут иметь повышенные АТл к рецепторам АЦХ. Примерно у 30% пострадавших подростков наблюдается повышение уровня АТл, но АТл к рецепторам АЦХ лишь изредка обнаруживаются в плазме крови детей препубертатного возраста. Некоторые из них с «-» титрами антихолинэстеразы имеют циркулирующие АТл к мышечно-специфической тирозинкиназе. Мышечно-специфическая тирозинкиназа локализуется в нервно-мышечном соединении и, по-видимому, необходима для в/утробного развития этого соединения. Дополнительными аутоАТл, связанными с иммунной миастении гравис, являются LRP4, титин, и АТл к рианодиновым рецепторы (RyR).
Младенцы, рожденные от матерей с миастенией, могут иметь ДН, неспособность сосать или глотать, а также генерализованную гипотонию и слабость, синдром, обычно называемый преходящей неонатальной миастенией. Они могут проявлять небольшую спонтанную двигательную активность в течение нескольких дней или недель. Возникновение симптомов обычно происходит в течение первых 1-3 дней жизни. Некоторые требуют ИВЛ и кормления через зонд в течение этого периода. Некоторым пациентам может потребоваться временное лечение пиридостигмина бромидом (ингибитором АХЭ). После того как аномальные АТл исчезают из крови и мышечной ткани, эти дети восстанавливают нормальную силу и не подвергаются повышенному риску развития миастении гравис в более позднем детстве.
Пациенты обычно имеют полное выздоровление к ~2 мес жизни. В редких случаях развивается последовательность акинезии плода с множественными контрактурами суставов (артрогрипоз), которые развиваются в/утробно из-за отсутствия движений плода. АТл к рецепторам АЦХ обычно м.б. обнаружены в материнской крови, но иногда материнские АТл могут и не обнаруживаться. Частота транзиторной неонатальной миастении, по оценкам, достигает 10-20% младенцев, рожденных от матерей с миастенией гравис.
в) Врожденные миастенические синдромы. Гетерогенная группа генетических заболеваний нервно-мышечной передачи в совокупности называется врожденными миастеническими синдромами. Этиология и патогенез этих синдромов не связаны ни с преходящей неонатальной миастенией, вызванной плацентарным переносом материнских АТл, ни с аутоиммунной миастенией гравис, несмотря на совпадение клинических симптомов. Врожденные миастенические синдромы почти всегда являются постоянными статическими расстройствами без спонтанной ремиссии (см. табл. 1 и табл. 2).
Распознается несколько разл. генетических форм, почти все с началом при рождении или в раннем младенчестве с симптомами, которые могут включать гипотонию, внешнюю офтальмоплегию, птоз, дисфагию, слабый крик, слабость лица, легкую мышечную усталость в целом, а иногда и ДН или нарушением дыхания, причем последняя часто сопровождается незначительной респираторной инфекцией. В детских формах часто встречаются такие симптомы, как утомляемость, задержка моторного развития и неустойчивые глазные симптомы (птоз и экстраокулярная мышечная слабость). Ингибиторы ХЭ оказывают благоприятное действие в большинстве случаев, но в некоторых формах симптомы и признаки действительно ухудшаются. Дети с большинством типов врожденной миастении гравис не испытывают миастенических кризов и редко имеют повышение уровня АТл к рецепторам АЦХ в плазме.
Мутации, ответственные за врожденные миастенические синдромы, идентифицированы в 24 разл. генах. Генетические мутации известны менее чем у половины детей с врожденным миастеническим синдромом. Наиболее распространенные гены, связанные с врожденными миастеническими синдромами, включают CHAT, CHRNE, DOK7, COLQ, GFPT и RAPSN. Врожденные миастенические синдромы м.б. вызваны мутациями, влияющими на белки, участвующие в синтезе АЦХ, слиянии везикул в синаптическую щель, распаде АЦХ в синаптической щели и обратном поглощении холина в субъединицах постсинаптического рецептора АЦХ, а также в путях постсинаптического гликозилирования. Белки, ассоциированные с базальной пластинкой, могут привести к аномалиям синаптической щели из-за мутаций в генах COLQ, COL13A1 и LAMB2.
Эти пути подчеркивают роль целостности белков внеклеточного матрикса в формировании и поддержании синапса. АТл к рецепторам АЦХ и мышечно-специфической тирозинкиназе обычно, но не всегда, отсутствуют в сыворотке крови, в отличие от аутоиммунных форм миастении гравис, поражающих детей старшего возраста и взрослых.
М.б. клинические признаки, помогающие в постановке диагноза (табл. 5) пациентам с эпизодами апноэ, напр. RAPSN, CHAT и COLQ. Эпизоды апноэ у пациентов с мутациями холинацилтрансферазы м.б. эпизодическими, но также бывают опасными для жизни. Хотя большинство врожденных миастенических синдромов передаются по наследству АуР-способом, есть несколько вариантов, где наследование происходит по Ауд-типу или Ауд-типу de novo, включая CHRNA1, CHRNB1, CHRND, CHRNE, и SYT2. Мутации в гене RAPSN могут привести к ранней гипотонии с ДН и эпизодическим апноэ, но могут также присутствовать в более мягких паттернах слабости конечностей с началом в детском или подростковом возрасте.
Гены, ассоциированные с более выраженным фенотипом миастенического синдрома конечностей, включают GFPT1, DPAGT1, ALG2, ALG14, GMPPB и PREPL. Гены, влияющие на постсинаптические субъединицы рецептора АЦХ, м.б. связаны с врожденным миастеническим синдромом медленных каналов, при котором пациенты могут иметь переменную слабость, как правило, с ухудшением с помощью ингибиторов АХЭ, а также врожденного миастенического синдрома быстрых каналов; они могут демонстрировать улучшение симптомов в ответ на ингибиторы боли.
г) Редкие другие причины миастении. Миастения гравис иногда ассоциируется с гипотиреозом, обычно с тиреоидитом Хашимото. Др. коллагеновые сосудистые заболевания, а также некоторые центронуклеарные миопатии могут быть связаны с дефектами нервно-мышечной передачи. Тимомы, отмеченные у некоторых взрослых, редко сосуществуют с миастенией гравис у детей. Точно так же карциномы легких, которые встречаются у взрослых, связанные с миастеническим синдромом Ламберта-Итона, не наблюдаются у детей. Синдром Ламберта-Итона у детей встречается редко, но сообщалось о лимфопролиферативных нарушениях и нейробластоме. Постинфекционная миастения гравис у детей носит преходящий характер и обычно следует за ветряной оспой — опоясывающей инфекцией на 2-5 нед в виде иммунного ответа.
д) Лабораторные исследования и диагностика. Миастения гравис — одно из немногих нервно-мышечных заболеваний, при которых ЭМГ является более специфичной диагностикой, чем биопсия мышц или нервов. Наблюдается сниженная реакция на повторяющуюся нервную стимуляцию; мышечные потенциалы быстро уменьшаются по амплитуде, пока мышца не становится невосприимчивой к дальнейшей стимуляции. Электрофизиологически эта реакция обусловлена уменьшением потенциалов концевых пластин с последующими повторяющимися стимулами, так что стимулы больше не приводят к потенциалам концевых пластин, которые достигают порога, приводящего к распространению потенциала двигательного действия. Это приводит к кумулятивному снижению амплитуды составного мышечного потенциала действия (СМАР) при повторных стимулах.
Снижение более чем на 10% между волнами 1:4 при повторной стимуляции диагностирует декрементальный ответ и наводит на мысль о нарушении нервно-мышечной передачи. Скорость проводимости двигательного нерва остается нормальной. Этот уникальный паттерн ЭМГ является электрофизиол. коррелятом утомляемости мышц и слабости, наблюдаемой клинически, и восстанавливается после введения ингибитора ХЭ. Миастенический декремент может отсутствовать или его трудно продемонстрировать в мышцах, которые не вовлечены клинически. Эта особенность может сбивать с толку в ранних случаях или у пациентов, демонстрирующих только слабость экстраокулярных мышц. Специальные электрофизиол. исследования необходимы при классификации врожденных миастенических синдромов и включают оценку количества рецепторов АЦХ на концевую пластинку и исследование функции концевой пластинки in vitro.
Эти специальные исследования и записи кинетических свойств каналов методом пэтч-кламп* выполняются на специальных биоптатах межреберных мышечных полосок, которые выполняются только в специализированных центрах.
P.S. * Пэтч-кламп — усовершенствованный электрофизиологический метод, который может непосредственно измерять мембранный потенциал, проходящий через клеточную мембрану. Если миастения ограничена экстраокулярной, леваторной и глоточной мышцами, повторяющаяся нервная стимуляция дистальных и проксимальных мышц (напр, abductor pollicis brevis muscle или trapezius muscle соответственно), хотя и диагностируется при генерализованном заболевании, обычно нормальна.
АТл против рецепторов АЦХ должны быть выявлены в плазме, но они возникают непоследовательно. АТл против рецептора мышечно-специфической тирозинкиназы следует искать у детей без циркулирующих АТл к рецепторам АЦХ, что является диагностическим признаком при повышенном уровне и дополнительно уточняет этиологию. Многие случаи врожденной миастении гравис являются результатом неспособности синтезировать или высвобождать АЦХ на пресинаптической мембране. В некоторых случаях ген, опосредующий фермент холинацетилтрансферазу для синтеза АЦХ, мутирует. В др. случаях наблюдается дефект в квантовом высвобождении везикул, содержащих АЦХ. Лечение таких больных ингибиторами ХЭ бесполезно. У некоторых пациентов, напр. с мутациями COLQ и DQK7, а также с миастенией медленных каналов ингибиторы АЦХ (напр., пиридостигмина бромид) могут не вызывать никакого ответа или ухудшения симптомов.
Клиническое генетическое тестирование врожденного миастенического синдрома может быть проведено с помощью панелей, которые являются коммерчески доступными и могут проводиться где угодно от 14 до 21 генов, ассоциированных с врожденными миастеническими синдромами.
Следует также искать др. серологические тесты аутоиммунного заболевания, такие как антинуклеарные АТл и аномальные иммунные комплексы. Если они «+», то вероятно наличие более общего аутоиммунного заболевания, включающего васкулит или ткани, отличные от мышц. Всегда следует проверять профиль ЩЖ. Уровень креатинкиназы в сыворотке крови нормальный при миастении гравис.
Сердце не задействовано, и данные ЭКГ остаются в норме. РОГК часто выявляют увеличение тимуса, но данная гипертрофия не является тимомой. Это может быть дополнительно определено с помощью томографии или КТ или МРТ переднего средостения, если рентгенографические данные неточны, но следует соблюдать осторожность при выборе оптимальных методов визуализации из-за радиационного воздействия при КТ и риска при проведении анестезии у пациента с миастенией, если необходима седативная МРТ.
Роль обычной мышечной биопсии при миастении гравис ограничена. В большинстве случаев это не требуется, но у 17% пациентов наблюдаются воспалительные изменения, иногда называемые лимфоизлияниями, которые интерпретируются некоторыми врачами как смешанное миастеническое полимиозитное иммунное расстройство. Мышечная биопсия ткани при миастении гравис показывает неспецифическую атрофию мышечных волокон II типа, аналогичную той, которая наблюдается при атрофии, стероидном воздействии на мышцы, ревматической полимиалгии и многих др. состояниях. Ультраструктура двигательных концевых пластин показывает упрощение мембранных складок; рецепторы АЦХ расположены в этих постсинаптических складках, как показывает бунгаротоксин (змеиный яд), который специфически связывается с рецепторами АЦХ.
Клиническим тестом на миастению гравис является введение ингибитора ХЭ короткого действия, обычно хлорида эдрофония. Птоз и офтальмоплегия уменьшаются в течение нескольких секунд, утомляемость др. мышц тоже уменьшается.
Рекомендации по применению ингибиторов холинэстеразы в качестве диагностического теста на миастении гравис у младенцев и детей:
1. Дети 2 лет и старше:
• У ребенка должна быть определенная утомляемая слабость, которую можно измерить, напр., птоз, дисфагия или неспособность шейных мышц поддерживать голову. Неспецифическая генерализованная слабость без двигательного дефицита ЧМН не является критерием.
• Следует начать в/в-инфузию, чтобы обеспечить возможность введения ЛС в случае возникновения нежелательной реакции.
• Во время теста рекомендуется проводить мониторинг ЭКГ.
• Доза атропина (атропина сульфата) (0,01 мг/кг) должна быть доступна в шприце, готовом к в/в-введению у постели больного во время эдрофониевого теста, чтобы блокировать острые мускариновые эффекты ингибитора ХЭ, главным образом спазмы в животе и/или внезапную диарею от повышенной перистальтики, обильные бронхотрахеальные выделения, которые могут затруднять проходимость ДП, либо, редко, сердечные аритмии. Некоторые врачи предварительно лечат всех пациентов атропином перед введением эдрофония, но это не рекомендуется, если нет в анамнезе реакции на тесты. Атропин может вызывать расширение зрачков в течение 14 дней после однократного приема, а зрачковый эффект гоматропина может длиться 4-7 дней.
• Хлорид эдрофония (Тенсилон) вводят в/в. Начальная тестовая доза составляет 0,01 мг/кг (для детей МТ<30 кг максНД 1 мг, а для детей МТ>30 кг максНД 2 мг. После начальной дозы можно вводить повторные дозы в/в. Для детей МТ<30 кг повторяют дозу по 1 мг каждые 30-45 с до максСД 5 мг. Для детей >30 кг повторяйте дозы по 1 мг каждые 30-45 с до максСД 10 мг. У взрослых средняя доза эдрофония для проявления «+» реакций составляет ~3,3 мг при птозе и ~2,6 мг при глазодвигательных симптомах. Побочные эффекты включают тошноту и рвоту; головокружение от брадикардии (антидот-атропин) и бронхоспазм — менее распространенные побочные эффекты. Тест на эдрофоний может проводиться в/м или п/к, но может потребоваться изменение дозировки.
• Эффекты должны быть видны в течение 10 с и исчезать в течение 120 с. Слабость измеряется, напр., расстоянием между верхним и нижним веками до и после введения ЛП, степенью внешней офтальмоплегии или способностью сделать глоток воды.
• Ингибиторы ХЭ длительного действия, такие как пиридостигмина бромид (Местинон), как правило, не так полезны для острой оценки миастенической слабости. Тест неостигмина (простигмина) может быть использован (как описано ниже), но может быть не таким окончательным диагностическим, как тест эдрофония.
2. Дети в возрасте до 2 лет:
• Младенцы в идеале должны иметь специфическую утомляемость и слабость, которую можно измерить, такую как птоз, дисфагия и неспособность шейных мышц поддерживать голову. Неспецифическая генерализованная слабость без двигательного дефицита ЧМН затрудняет оценку результатов, но иногда может быть критерием.
• В/в-доступ для ЛС в случае неблагоприятного эффекта испытуемого ЛП.
• Во время теста рекомендуется проводить мониторинг ЭКГ.
• Предварительное лечение атропином (атропина сульфатом) для блокирования мускариновых эффектов исследуемого ЛП не рекомендуется, но атропин (атропина сульфат) должен быть доступен у постели больного в подготовленном шприце. Его следует вводить в/в 0,01 мг/кг PRN.
• Эдрофрниум не рекомендуется применять у младенцев; его действие слишком кратковременно для объективной оценки, и сообщается о повышенной частоте острых сердечных аритмий у младенцев, особенно новорожденных, получающих этот ЛП.
• Простигмина метилсульфат (неостигмин) вводят в/м 0,04 мг/кг. Если результат «-» или двусмысленный, то через 4 ч после первой дозы можно ввести еще одну дозу 0,04 мг/кг (обычно 0,5-1,5 мг). Пик эффекта наблюдается через 20-40 мин. В/в-введение простигмина противопоказано из-за риска развития сердечных аритмий, включая фатальную фибрилляцию желудочков, особенно у маленьких детей.
• Ингибиторы ХЭ длительного действия, вводимые внутрь, такие как пиридостигмина бромид (Местинон), как правило, не так полезны для острой оценки миастенической слабости, поскольку начало и продолжительность заболевания менее предсказуемы.
• Тест должен проводиться в ОНМП, больничной палате или ОРИТ; важным вопросом является подготовка к возможным осложнениям, таким как сердечная аритмия или холинергический криз, как описано выше.
е) Лечение. Некоторые пациенты с легкой формой миастении гравис не нуждаются в лечении. Основными терапевтическими ЛС являются ЛП, ингибирующие ХЭ. Пиридостигмина бромид (Местинрн) можно вводить внутрь, с 0,5-1 мг/кг Q4-6H, пока пациент бодрствует, максД 60 мг. МаксСД составляет 7 мг/кг/сут, причем большинство взрослых достигают эффекта при максСД <960 мг/сут, разделенной на 4-8 приемов. Пиридостигмина бромид назначается в форме короткого действия, а также может быть использован в форме длительного действия перед сном для пациентов с большей слабостью при пробуждении утром.
Передозировка ингибиторов ХЭ вызывает холинергические кризы с такими симптомами, как повышенная секреция, диарея и судороги; атропин блокирует мускариновые эффекты, но не блокирует никотиновые эффекты, которые вызывают дополнительную слабость скелетных мышц.
При редкой семейной миастении гравис, вызванной отсутствием концевой пластинки АХЭ, ингибиторы ХЭ не помогают и часто вызывают повышенную слабость; этих пациентов можно лечить эфедрином или диаминопиридином, оба из которых увеличивают высвобождение АЦХ из концевых аксонов.
Из-за аутоиммунной основы заболевания длительное стероидное лечение преднизоном м.б. эффективным. Тимэктомия должна быть рассмотрена и может обеспечить излечение. Тимэктомия наиболее эффективна у пациентов с высокими титрами АТл к рецепторам АЦХ в плазме крови и симптоматикой в течение <2 лет. Тимэктомия неэффективна при врожденных и семейных формах миастении гравис. Лечение гипотиреоза обычно устраняет связанную с ним миастению без применения ингибиторов ХЭ или стероидов.
Если специфическая генетическая мутация может быть идентифицирована у пациента с одним из врожденных миастенических синдромов, для некоторых из них доступны специфические терапевтические подходы, которые отличаются от перечисленных выше методов лечения.
Плазмаферез является эффективным методом лечения у некоторых детей, особенно у тех, кто не реагирует на стероиды, но плазмообменная терапия обеспечивает лишь временную ремиссию. В/в-иммуноглобулин полезен, и его следует попробовать перед плазмаферезом, потому что он менее инвазивен. Плазмаферез и в/в-введение Ig, по-видимому, наиболее эффективны у пациентов с высоким уровнем циркулирующих АТл к рецепторам АЦХ. Пациенты трудноподдающиеся лечению, а также пациенты с миастенией гравис, связанной с мышечно-специфической тирозинкиназой, могут более эффективно реагировать на ритуксимаб, моноклональные АТл к в/клеточному АГн CD20.
Новорожденные с преходящей материнской миастенией гравис нуждаются в ингибиторах ХЭ только в течение нескольких дней или иногда в течение нескольких недель, особенно для того, чтобы позволить кормление. Никакого др. лечения обычно не требуется. При врожденной миастении гравис, не передающемся от матери, идентификация специфического молекулярного дефекта важна для лечения; в табл. 6 обобщены конкретные методы лечения для каждого типа.
ж) Осложнения. Дети с миастенией гравис не переносят нервно-мышечные блокирующие ЛП, такие как сукцинилхолин и панкурония бромид, м.б. парализованы в течение нескольких недель после однократного приема. Анестезиолог должен тщательно осмотреть пациентов с миастенией, которым требуется хирургическая анестезия, и такие анестетики должны назначаться только опытным врачом/анестезиологом. Кроме того, некоторые АБ могут усиливать миастению, и их следует избегать; к ним относятся аминогликозиды, бета-блокаторы, прокаинамид, хлорохин и фторхинолоны.
з) Прогноз. Некоторые пациенты с аутоиммунной миастенией гравис испытывают спонтанную ремиссию через несколько месяцев или лет; др. имеют постоянное заболевание, распространяющееся во взрослую жизнь. Иммуносупрессия, тимэктомия и лечение сопутствующего гипотиреоза могут обеспечить излечение. Генетически детерминированные врожденные миастенические синдромы могут иметь первоначальное ухудшение в младенчестве, но затем оставаться статичными на протяжении всего детства и во взрослой жизни.
и) Другие причины нервно-мышечной блокады. ФОС хим. в-ва, обычно используемые в качестве инсектицидов, могут вызывать миастенический синдром у детей, подвергающихся воздействию этих токсинов.
Ботулизм возникает в результате приема пищи, содержащей токсин Clostridium botulinum, гр/п, спороносной анаэробной бациллы. Инкубационный период короткий, всего несколько часов, и симптомы начинаются с тошноты, рвоты и диареи. Вскоре следует вовлечение ЧМН с диплопией, дисфагией, слабым сосанием, слабостью лица и отсутствием рвотного рефлекса. Механизм заключается в расщеплении ботулиническим токсином нескольких структурных гликопротеинов стенки (т.е. мембраны) синаптических везикул в аксональных терминалях. Эти гликопротеины включают синаптобревин и синаптотагмин, но синаптофизин устойчив.
При инфантильном ботулизме, который классически проявляется в возрасте от 4 до 7 мес, мёд, а также споры из грязи (напр., вблизи строительных площадок) являются распространенными источниками загрязнения. Самыми ранними признаками обычно являются запор, нарушение аппетита, а затем слабый крик. При осмотре у пациентов гипотония, слабость мимических мышц, дисфагия и слабый рвотный позыв. Может возникнуть генерализованная слабость с риском ДН. Затем развиваются генерализованная гипотония и слабость, которые могут прогрессировать до ДН. Нервно-мышечная блокада документируется ЭМГ с повторяющейся стимуляцией нервов.
Медленная повторяющаяся нервная стимуляция может показать декрементный ответ, а исходные амплитуды СМАР м.б. низкими. При быстрой повторяющейся стимуляции нервов возникает постепенная реакция. Исследования ЭМГ/повторной стимуляции нервов могут помочь в подтверждении диагноза, если клиническая картина не является однозначной. Однако при подозрении на ботулинический токсин исследования следует провести исследование стула пациента, а затем как можно скорее начинать лечение ботулиническим иммуноглобулином IV (Baby-BIG или BIG IV). BIG-IV, который представляет собой АТл к токсину ботулизма человеческого происхождения, одобрен FDA США для лечения детского ботулизма типов А и В.
Раннее использование BIG-IV сократило общую продолжительность госпитализации и уменьшило время до выздоровления. Дыхательная и нутритивная поддержка может потребоваться в течение нескольких дней или недель, пока токсин не будет выведен из организма.
Клещевой паралич — нарушение высвобождения АЦХ из аксональных окончаний из-за нейротоксина, блокирующего деполяризацию. Он также поражает крупные миелинизированные двигательные и сенсорные нервные волокна. Этот токсин вырабатывается древесным клещом или собачьим клещом, насекомыми, распространенными в Аппалачах и Скалистых горах Северной Америки. Клещ внедряет свою головку в кожу, обычно в кожу головы, и максимальная выработка нейротоксина через ~5-6 дней. Двигательные нарушения включают слабость, потерю координации и иногда восходящий паралич, напоминающий синдром Гийена-Барре. Сухожильные рефлексы утрачены. Сенсорные симптомы покалывания, парестезии могут возникать на лице и конечностях.
Диагноз подтверждается идентификацией клеща, а лечение предполагает оперативное удаление всего клеща. Важно внимательно следить за пациентами, потому что у некоторых пациентов может наблюдаться ухудшение симптомов в течение первых суток после удаления клеща. У большинства пациентов наблюдается быстрое улучшение в течение нескольких часов или нескольких дней с момента удаления клеща.