Наследственные моторно-сенсорные невропатии представляют собой группу прогрессирующих заболеваний периферических нервов (табл. 1).
Двигательные компоненты обычно доминируют в клинической картине, но сенсорные и вегетативные симптомы появляются позже. Биопсия сурального нерва раньше считалась наиболее точным средством диагностики, но с расширением знаний молекулярной генетики этой группы заболеваний диагноз большинства из них м.б. подтвержден менее инвазивным генетическим тестированием.
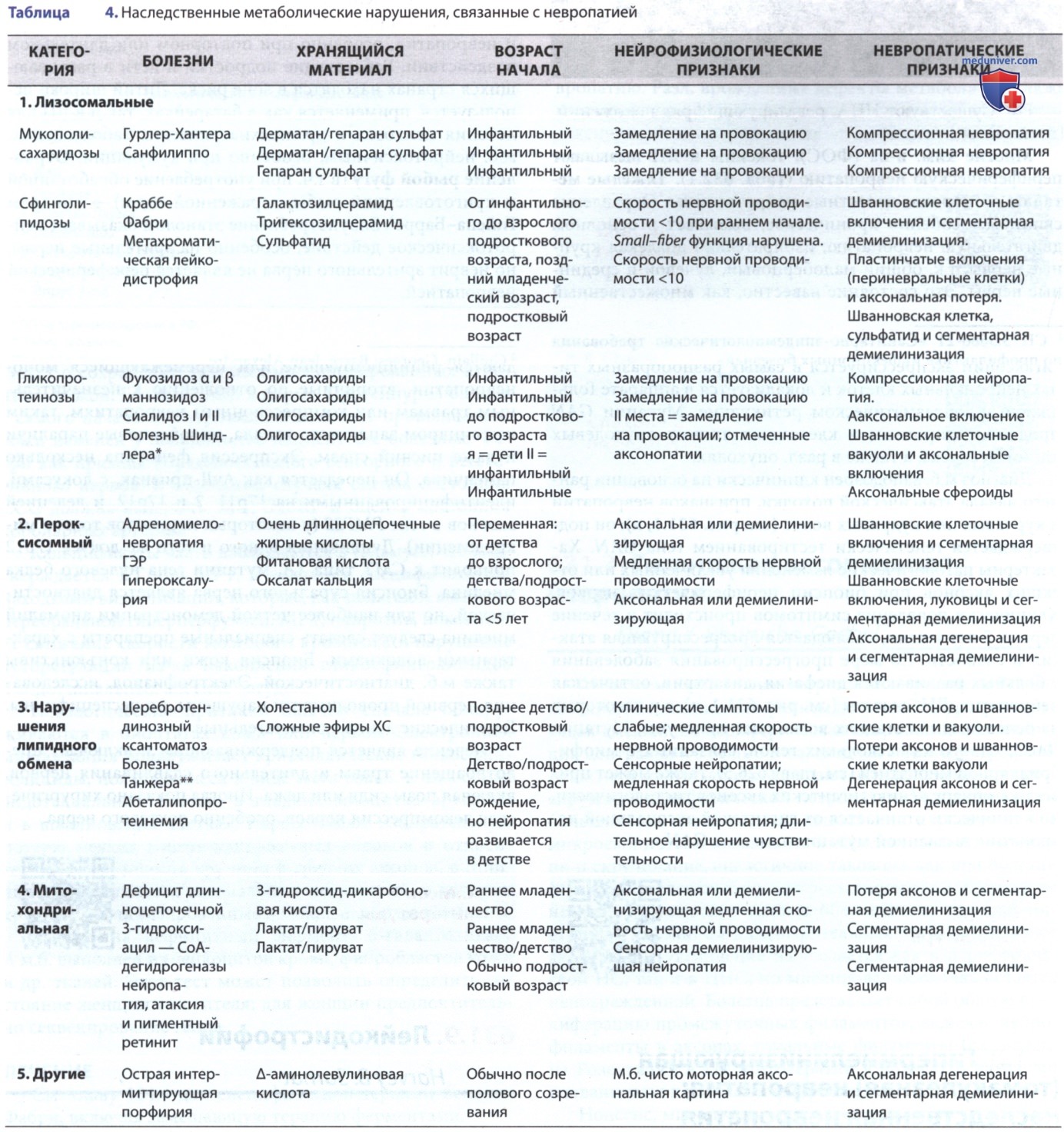
ЭМГ остается полезным дополнением в клинической диагностике и помогает различать демиелинизирующую или гипомиелинизирующую и аксональную формы. Клинические признаки отмечены в табл. 2 и 3.
Классификация наследственных моторно-сенсорных невропатий сложна тем, что ни одна простая унифицирующая схема не способна охватить все клинические проявления и перекрывающиеся генетические особенности (см. табл. 1). При некоторых невропатиях разнообразный генотип мутаций разл. генов в разных хромосомных локусах может приводить к сходному фенотипу. Представляем одну из классификаций.
I. Наследственные невропатии, вторичные по отношению к общим заболеваниям.
III. Синдромные невропатии, включая врожденные гипомиелинизирующие.
IV. Наследственная сенсорная невропатия (болезнь Рефсума (Refsum, Sigvald Bernhard)).
а) Малоберцовая мышечная атрофия (болезнь Шарко-Мари-Тута, наследственная моторно-сенсорная невропатия с ранним началом типа IIA). Болезнь Шарко-Мари-Тута (СМТ) — наиболее распространенная генетически детерминированная невропатия, имеет общую распространенность 3,8:100 000 населения. Она передается как АуД-признак с выраженностью 83%; локус 17р11.2 является местом аномального гена.
АуР-передача также описана, но встречается реже. Генным продуктом является периферический миелиновый белок 22 (РМР22). Гораздо более редкая Х-сцепленная наследственная моторно-сенсорная невропатия с ранним началом типа I является результатом дефекта в локусе Xq13.1, вызывающего мутации в белке gap — соединения коннексин-32. Сообщалось и о др. формах (см. табл. 1).
Мутации влияют как на связывание жирных кислот РМР22, так и на кинетику его мембранных взаимодействий.
1. Клинические проявления. Большинство пациентов не имеют симптомов до позднего детства или раннего подросткового возраста, но у маленьких детей иногда проявляются нарушения походки уже на 2-м году жизни. Малоберцовый и большеберцовый нервы поражаются раньше и тяжелее. Детей с этим расстройством часто описывают как неуклюжих, легко падающих или спотыкающихся о собственные ноги. Применение инструмента нестабильности голеностопного сустава Камберленда (Cumberland) является средством объективного документирования и отслеживания этой симптоматики. Появление симптомов м.б. отложено до 5-й декады жизни.
Мышцы переднего отдела голеней истощаются, и ноги имеют характерный аистоподобный контур. Мышечная атрофия сопровождается прогрессирующей слабостью дорсифлексии голеностопного сустава и последующим провисанием стопы. Процесс двусторонний, но м.б. слегка асимметричным. PES cavus деформации неизменно развиваются в результате денервации внутренних мышц стопы, что еще больше дестабилизирует походку. Атрофия мышц предплечий и кистей обычно не так сильна, как у нижних конечностей, но в запущенных случаях контрактуры запястий и пальцев приобретают вид когтистой руки.
Слабость проксимальных мышц является поздним признаком и обычно протекает в легкой форме. Аксиальные мышцы не страдают.
Болезнь медленно прогрессирует на протяжении всей жизни, но у пациентов иногда наблюдается ускоренное ухудшение функции в течение нескольких лет. Большинство пациентов могут себя обслуживать и имеют нормальную продолжительность жизни, хотя для стабилизации лодыжек требуются ортопедические приспособления.
Сенсорное нарушение в основном затрагивает крупные миелинизированные нервные волокна, которые передают проприоцептивную информацию и вибрационные ощущения, но порог боли и температуры также может увеличиваться. Некоторые дети жалуются на покалывание или жжение в ногах, но боль бывает редко. Поскольку мышечная масса уменьшается, нервы более уязвимы к травмам или сжатию. Вегетативные симптомы могут выражаться в виде плохого вазомоторного контроля с пятнами или бледностью кожи стоп и неадекватно холодными стопами.
Нервы часто становятся ощутимо увеличенными. Рефлексы с сухожилий исчезают в дистальных отделах. ЧМН не поражены. Контроль сфинктера остается хорошо сохраненным. Вегетативная нейропатия не влияет на ССС, ЖКТ или МВП. Интеллект в норме. Уникальная точечная мутация в РМР22 вызывает прогрессирующую сенсоневральную тугоухость, но это обычно происходит позже, чем периферическая нейропатия.
Синдром Давиденкова — это вариант наследственной моторно-сенсорной невропатии с ранним началом I типа со скапулоперонеальным распределением.
2. Лабораторные исследования и диагностика. Скорость моторной и сенсорной нервной проводимости значительно снижается, иногда до 20% нормального времени проводимости. В случаях отсутствия семейного анамнеза следует обследовать обоих родителей и провести исследования нервной проводимости.
ЭМГ и биопсия мышц обычно не требуются для диагностики, но они свидетельствуют о многих циклах денервации и реиннервации. Уровень КФК в сыворотке крови в норме. Белок СМЖ м.б. повышен, но клетки в СМЖ не появляются.
Биопсия сурального нерва необходима для диагностики. Крупные и средние миелинизированные волокна уменьшаются в количестве, коллаген увеличивается, а характерные луковичные образования пролиферированной цитоплазмы шванновских клеток окружают аксоны. Эта патологическая находка называется интерстициальной гипертрофической невропатией. Также происходит обширная сегментарная демиелинизация и ремиелинизация. Окончательный молекулярно-генетический диагноз м.б. поставлен при исследовании крови.
3. Лечение. Стабилизация лодыжек является основной проблемой. На ранних стадиях часто достаточно жестких ботинок, которые доходят до середины голени, особенно когда пациенты ходят по неровным поверхностям, таким как лед, снег или камни. По мере того как дорсифлексоры лодыжек еще больше ослабевают, легкие пластиковые шины м.б. изготовлены на заказ, которые проходят под стопой и вокруг задней части лодыжки. Их носят внутри носков, и они не видны, что очень важно психологически. Могут потребоваться наружные короткие распорки для ног, когда нога будет полностью выпадать. В некоторых случаях м.б. рассмотрено хирургическое сращение голеностопного сустава.
Нога должна быть защищена от травматических повреждений. В запущенных случаях компрессионная невропатия во время сна м.б. предотвращена путем размещения мягких подушек под или между голенями. Жгучие парестезии стоп встречаются редко, но часто устраняются фенитоином, карбамазепином или габапентином. Прогрессивное упражнение на сопротивление для дорсифлексии стопы может ослабить прогрессирование слабости.
Наследственная сенсорная вегетативная невропатия I в предварительных исследованиях лечилась пероральным L-серином с биохим. улучшениями (снижением токсичных метаболитов).
б) Малоберцовая мышечная атрофия (аксональный тип). Малоберцовая мышечная атрофия клинически сходна с наследственной моторно-сенсорной невропатией с ранним началом I типа, но скорость прогрессирования медленнее и инвалидизация меньше. ЭМГ обнаруживает денервацию мышц. Биопсия сурального нерва выявляет аксональную дегенерацию, а не демиелинизацию и завитки шванновских клеточных процессов, типичных для типа I. Локус находится на хромосоме 1 в 1р35-р36; это не наследственная моторно-сенсорная невропатия с ранним началом типа I, хотя оба заболевания передаются как АуД-признаки.
АуР инфантильная моторная аксональная невропатия может значительно имитировать инфантильную СМА.
в) Врожденная гипомиелинизирующая невропатия и болезнь Дежерина-Сотта (наследственная моторносенсорная невропатия с ранним началом III типа). Врожденная гипомиелинизирующая невропатия — интерстициальная гипертрофическая невропатия с АуД-передачей, клинически сходная с наследственной моторно-сенсорной невропатией с ранним началом I типа, но более тяжелая. Симптомы развиваются в раннем младенчестве и быстро прогрессируют, сопровождаясь гипотонией, затруднением дыхания и питания.
Аномалии зрачков, такие как отсутствие реакции на свет и зрачок Аргайлла Робертсона (Robertson, Argyll), встречаются довольно часто. Кифосколиоз и деформации в виде высокого свода стопы развиваются в 35% случаев. Нервы становятся ощутимо вовлеченными в раннем возрасте. Болезнь Дежерина-Сотта — медленно прогрессирующий вариант, начинающийся обычно в возрасте до 5 лет.
АуР-форма врожденной гипомиелинизирующей невропатии также известна и м.б. вызвана различными генетическими мутациями, включая MTMR2, РМР22, EGR2 и MPZ. Вторичная мутация в гене EGR2 может усилить клиническую манифестацию болезни Дежерина-Сотта. Неонатальная гипотония и задержка развития в младенчестве являются характерными клиническими признаками. Многие пациенты проявляют врожденную нечувствительность к боли. ЧМН страдают непоследовательно, а ДН и дисфагия являются редкими осложнениями. Сухожильные рефлексы отсутствуют. Артрогрипоз обнаруживают при рождении по крайней мере в половине случаев.
Луковичные образования, видимые в образце биопсии сурального нерва, ярко выражены. Происходит также гипомиелинизация. При рецессивной форме гипомиелинизация не во всех случаях может сопровождаться интерстициальной гипертрофией.
Генетический локус 17р11.2 идентичен локусу наследственной моторно-сенсорной невропатии с ранним началом типа I или СМТ. Моноаллельные мутации в MPZ (myelin protein zero), РМР22 или EGR2 (early grow response 2) являются наиболее частыми генетическими причинами. Клинические и патологические различия м.б. фенотипическими вариантами одного и того же заболевания, аналогично ситуации при мышечных дистрофиях Дюшенна (Duchenne, Guillaume) и Беккера (Becker, Peter Emil). АуР-форма болезни Дежерина-Сотта не полностью документирована.
г) Синдром Русси-Леви. Синдром Русси-Леви (Roussy, Gustave; Levy, Gabrielle) определяется как сочетание наследственной моторно-сенсорной невропатии с ранним началом II типа и мозжечкового дефицита, напоминающего атаксию Фридрейха (Friedreich, Nikolaus), но при этом без кардиомиопатии.
д) Болезнь Рефсума (наследственная моторно-сенсорная невропатия с ранним началом типа IV) и инфантильная болезнь Рефсума. Болезнь Рефсума (Refsum, Sigvald Bernhard) — редкое АуР-заболевание, вызванное ферментативным блоком β-окисления фитановой кислоты в пристановую. Фитановая кислота — жирная кислота с разветвленной цепью, получаемая в основном из пищевых источников: шпината, орехов и кофе. Уровень фитановой кислоты значительно повышен в плазме, СМЖ и мозговой ткани. Фитановые и длинноцепочечные жирные кислоты м.б. липотоксичными, нарушая функцию митохондрий в ЦНС и периферической НС.
В СМЖ наблюдается альбуминоцитологическая диссоциация с концентрацией белка 100-600 мг/дл. Исследования генетических связей идентифицируют два разл. локуса в 10р13 и 6q22-q24 с генетическими мутациями PHYH и РЕХ7 соответственно. Инфантильная форма также м.б. вызвана генами РЕХ1, РЕХ2 или РЕХ26, которые производят как клинические, так и биохим. отличия от классической формы и включают незначительный дисморфизм лица, пигментный ретинит, нейросенсорную тугоухость, гиперхолестеринемию, гепатомегалию. Накопление фитановой кислоты при инфантильной ГЭР вторично по отношению к первичному пероксисомному расстройству; следовательно, АуР ГЭРБ действительно др. заболевание.
Клиническое начало классической ГЭРБ обычно наступает в возрасте 4-7 лет с периодической моторной и сенсорной нейропатией. В разл. степени развиваются также атаксия, прогрессирующая нейросенсорная тугоухость, пигментный ретинит с потерей ночного зрения, ихтиоз, дисфункция печени. Скелетные пороки развития с рождения и находки в ССС в виде нарушений проводимости и кардиомиопатии появляются у большинства пациентов. Скорость моторной и сенсорной нервной проводимости замедляется. Биопсия сурального нерва выявляет потерю миелинизированных аксонов.
Лечение заключается в диетическом питании и периодическом плазмоферезе. При правильном ведении продолжительность жизни м.б. нормальной. Потеря слуха из-за поражения слухового нерва иногда м.б. улучшена с помощью кохлеарной имплантации.
е) Болезнь Фабри. Болезнь Фабри (Fabry, Johannes), редкий Х-сцепленный рецессивный признак, приводит к накоплению церамид-тригексозида из-за дефицита фермента церамид-тригексозидазы, который расщепляет терминальную галактозу от церамидтригексозида (церамид-глюкоза-галактоза-галактоза), что приводит к накоплению этого тригексозидного липида в тканях нейронов ЦНС, шванновских, периневральных, ганглиозных клетках мезентериального сплетения, кожи, почек, эндотелия кровеносных сосудов и гладких мышц, клетках ССС, потовых желез, роговицы и костного мозга. Это происходит в результате миссенс-мутации, нарушающей кристаллографическую структуру α-галактозидазы А.
1. Клинические проявления. Первые проявления отмечаются в позднем детском или подростковом возрасте, с повторяющимися эпизодами жгучей боли и парестезиями стоп и голеней, настолько сильными, что пациенты не могут ходить. Эти эпизоды часто вызываются лихорадкой или ФН. Объективные сенсорные и моторные нарушения при неврологическом обследовании не выявляются, рефлексы сохраняются. Поражение вегетативных нервов является почти универсальным и может вызвать нарушения сердечного ритма, кожные реакции и нарушения перистальтики ЖКТ, но вегетативная экспрессия варьирует у разных пациентов.
Поражение ССС не ограничивается вегетативными нарушениями — аритмиями и дефектами проводимости, а может также включать гипертрофию ЛЖ, ИБС и клапанную инфильтративную миопатию. Характерные поражения кожи наблюдаются в области промежности, мошонки, ягодиц и периумбиликальной зоны в виде плоских или приподнятых красно-черных телеангиэктазий, известных как angiokeratoma corporis diffusum. М.б. гипогидроз. Помутнение роговицы, катаракта и некроз головок бедренных костей — непостоянные признаки. Могут возникать извитости сосудов сетчатки, позвоночных и базилярных артерий. Заболевание носит прогрессирующий характер.
АГ и ОПН обычно откладываются до ранней взрослой жизни. Повторяющиеся инсульты возникают в результате поражения сосудистой стенки. Смерть часто наступает в 5-м десятилетии вследствие инфаркта ГМ или ОПН, но выраженная симптоматика возникает уже в детском возрасте, несмотря на отсутствие серьезной органной недостаточности. Гетерозиготные носители женского пола м.б. бессимптомными или с мало выраженными симптомами, чем мужчины; помутнение роговицы составляет 70-80%, хотя катаракта встречается редко.
2. Лабораторные результаты. Скорость проводимости двигательных и сенсорных нервов нормальная и лишь слегка замедлена, что свидетельствует о сохранении крупных миелинизированных нервных волокон. Белок в СМЖ в норме. Протеинурия бывает с самого начала. Электрохимический тест проводимости кожи изменен у большинства пациентов с болезнью Фабри как признак поражения малого сенсорного и вегетативного нервов. В отдельных случаях оценка состояния ССС должна включать ЭКГ, ЭхоКГ и оценку состояния коронарных артерий.
Кальцификаты часто наблюдаются в таламусе, что подтверждается КТ или МРТ, и являются специфическими находками визуализации, которые, как полагают, вызваны церебральной гиперперфузией. МРТ, напротив, показывает снижение скорости мозгового кровотока и нарушение ауторегуляции из-за накопления гликосфинголипидов в эндотелиальных клетках сосудов.
Патологические признаки обычно сначала обнаруживаются в биоптатах кожи или нервов. Электронная микроскопия обнаруживает кристаллические гликосфинголипиды, появляющиеся в виде zebra bodies, в лизосомах эндотелиальных клеток, в гладких миоцитах артериол и в шванновских клетках. Нервы имеют избирательную потерю мелких миелинизированных волокон и относительную сохранность крупных и средних аксонов, в отличие от большинства аксональных невропатий, в которых крупные миелинизированные волокна наиболее изменены.
Анализ на дефицитный фермент, а-галактозидазу А м.б. выполнен из лейкоцитов крови, фибробластов кожи и др. тканей. Этот тест может позволить определить состояние женщины-носителя; для женщин предпочтительно секвенирование генов.
3. Лечение. См. отдельную статью на сайте для специфической терапии болезни Фабри, включая замещающую терапию ферментами.
Медикаментозная терапия невропатий включает лечение инициирующего заболевания и терапию, направленную на невропатическую боль независимо от этиологии. Боль м.б. жгучей или связанной с парестезией, гипералгезией (ненормальной реакцией на вредные раздражители) или аллодинией (вызванной вредными раздражителями). Невропатическая боль часто успешно справляется с помощью ТЦА; CJ4O3C менее эффективны. Эффективны противосудорожные ЛС (карбамазепин, фенитоин, габапентин, ламотриджин), а также наркотические и ненаркотические анальгетики. Ферментная заместительная терапия улучшила краткосрочный и долгосрочный прогноз клинической невропатии, а также снизила повышенную скорость кровотока в ГМ.
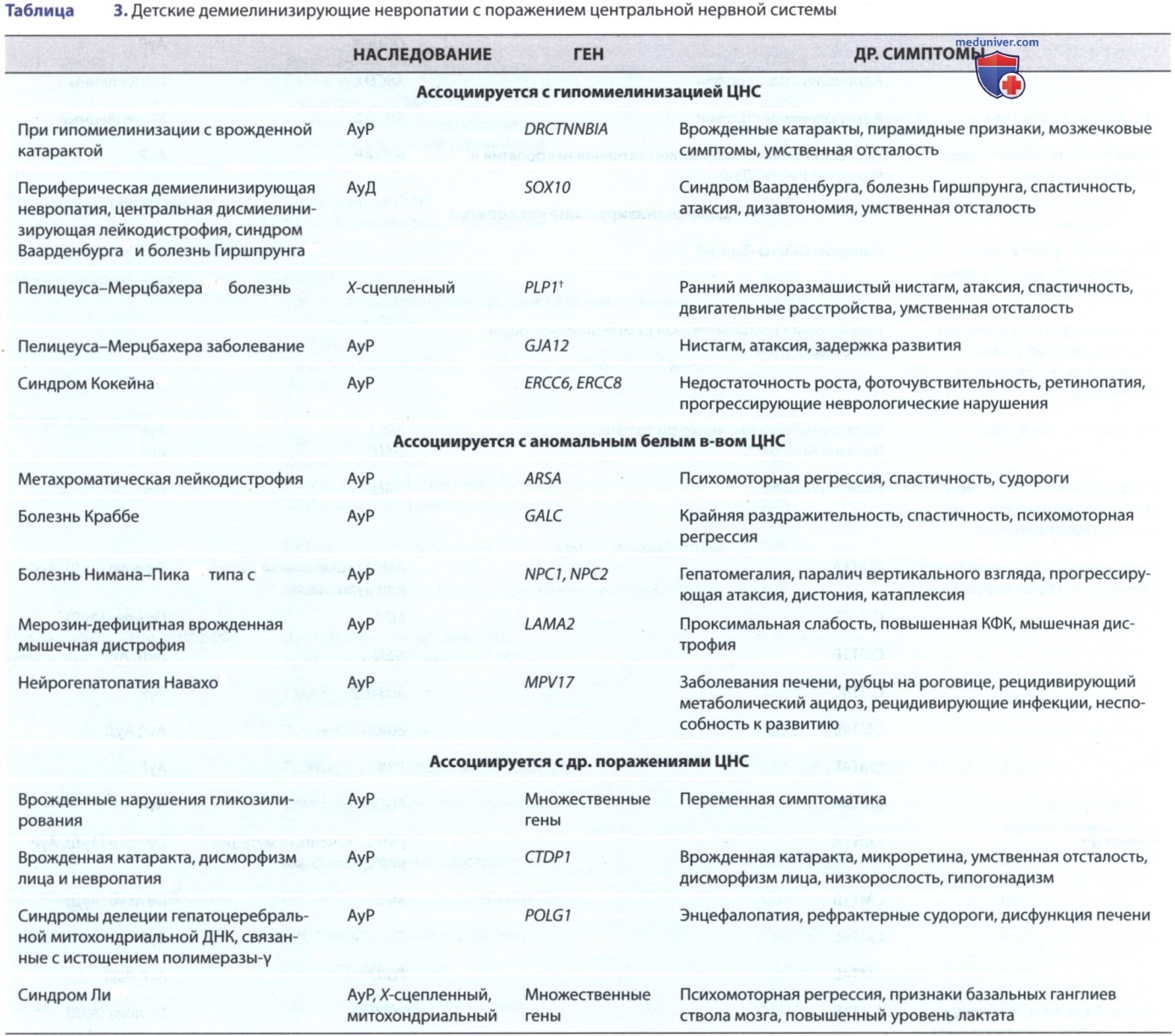
ж) Гигантская аксональная невропатия. Гигантская аксональная невропатия — редкое АуР заболевание, начинающееся в раннем детстве. Это прогрессирующая смешанная периферическая невропатия и дегенерация центрального белого в-ва, сходная с лейкодистрофией. Атаксия и нистагм сопровождаются признаками прогрессирующей периферической невропатии. У подавляющего большинства детей волосы вьются или курчавятся, что микроскопически показывает изменение диаметра стержня и скручивание, аналогичное таковому как при болезни Менкеса (Menkes, John Hans); следовательно, микроскопическое исследование нескольких волосков головы обеспечивает простой инструмент скрининга в подозрительных случаях.
Очаговое аксональное увеличение наблюдается как в периферической НС, так и в ЦНС, но миелиновая оболочка остается неповрежденной. Болезнь представляет собой общую пролиферацию промежуточных филаментов, включая нейрофиламенты в аксонах, глиальные филаменты (т.е. волокна Розенталя (Rosenthal)) в ГМ, цитокератин в волосах и виментин в шванновских клетках и фибробластах.
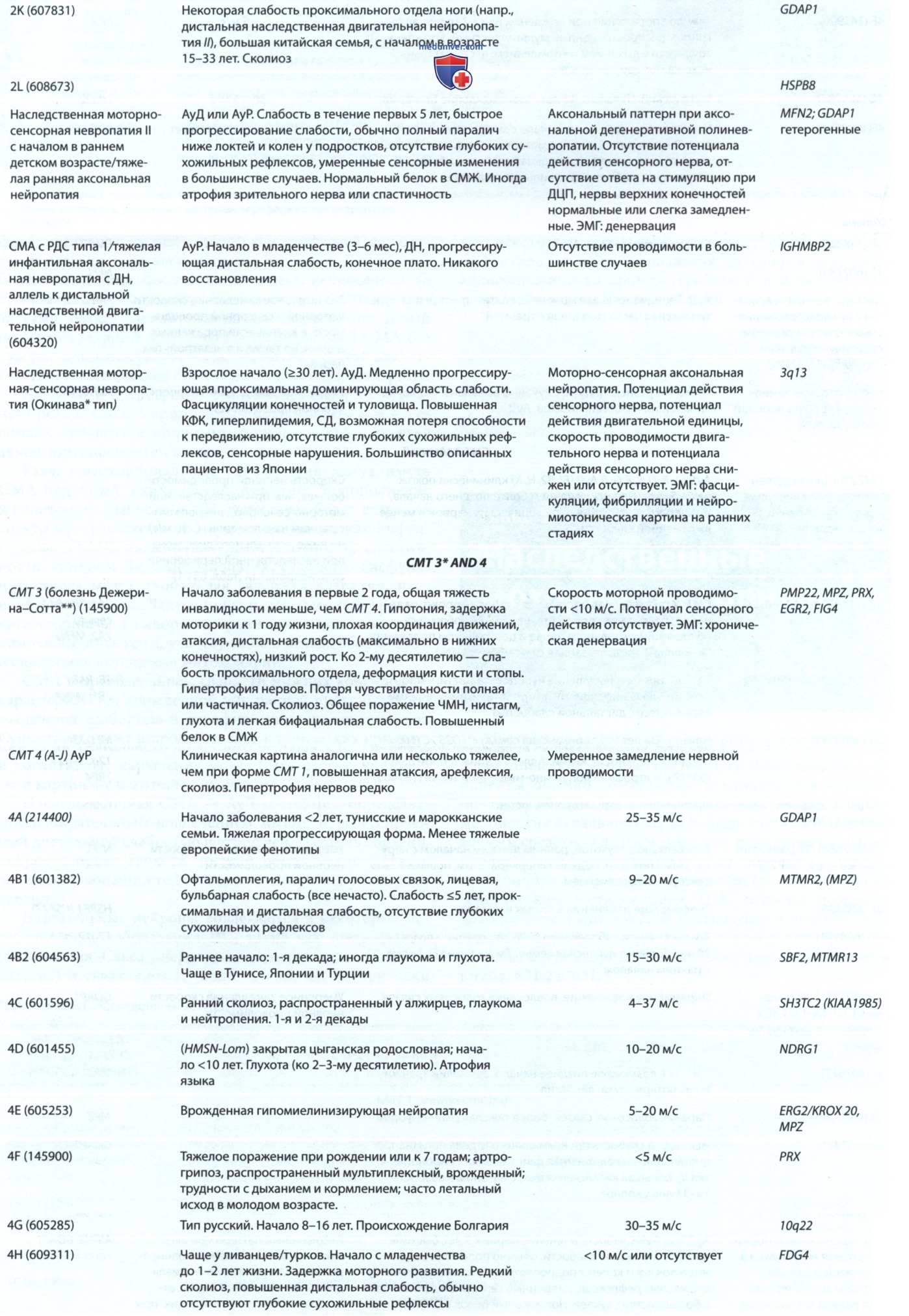
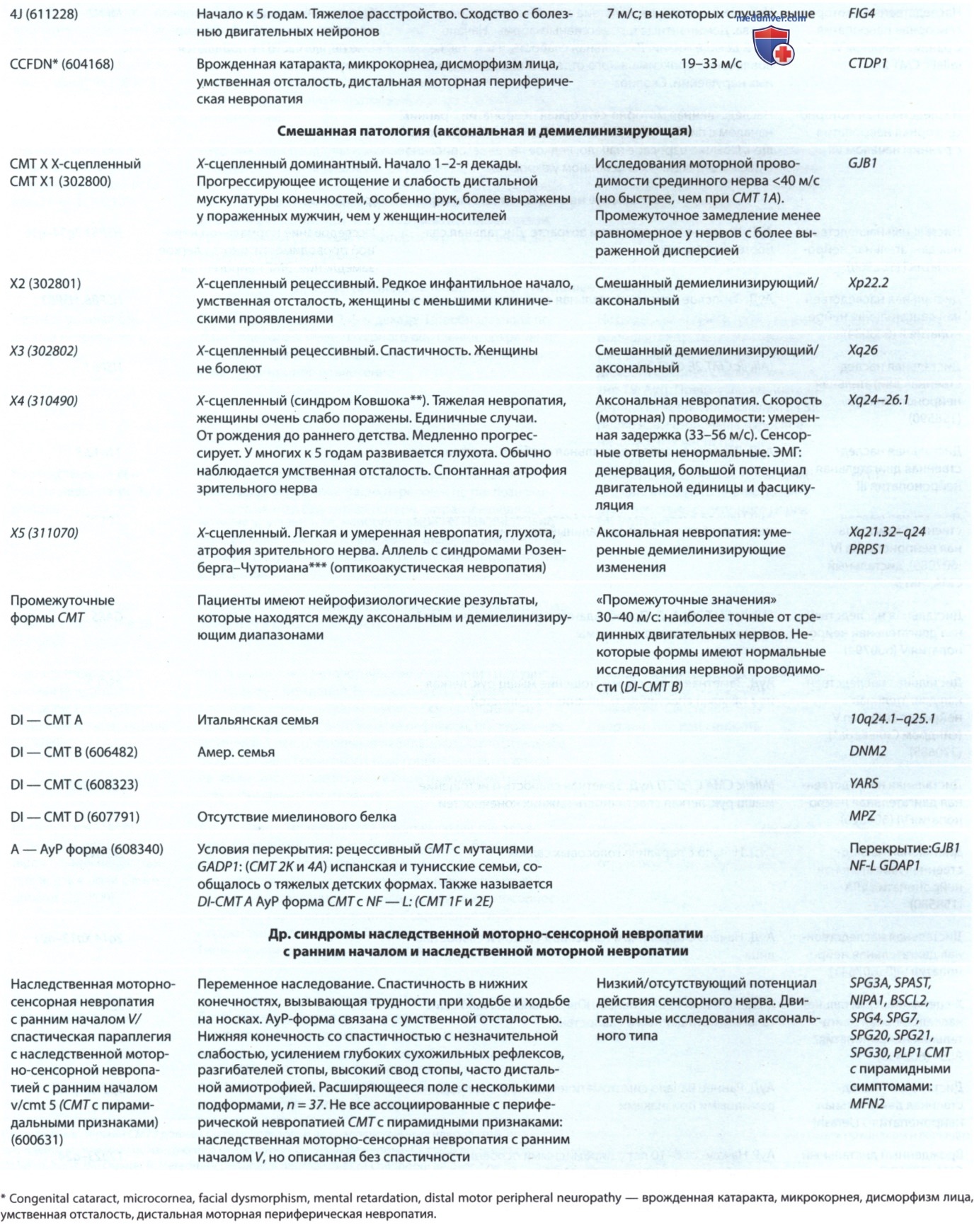
Нонсенс, миссенс-мутации, мутации сайта сплайсинга или делеции происходят в гене GAN с аллельной гетерогенностью в 16q24. Эти мутации ответственны за дефектный синтез белка гигаксонина, члена цитоскелетного суперсемейства BTB/kelch, имеющего решающее значение для связи между промежуточными белками и клеточной мембраной. МРТ показывает поражения белого в-ва ГМ, похожие на лейкодистрофии (рис. ниже), А MP-спектроскопия выявляет повышенное соотношение холин/креатин и миоинозитол/креатин при снижении N-ацетил аспартата, что указывает на демиелинизацию и глиальную пролиферацию, а также на потерю аксонов.
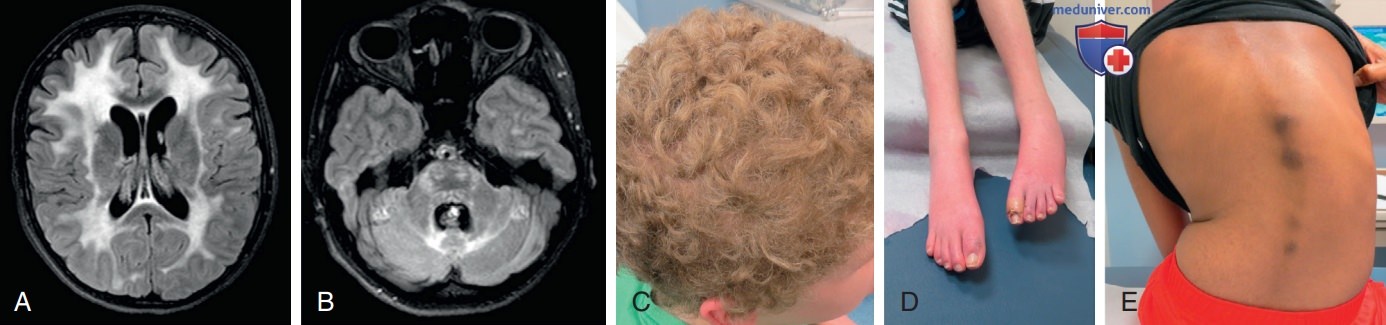
Гигантская аксональная невропатия: A, B — аномалии белого в-ва на последовательностях T2/FLAIR, включающие ствол головного мозга и мозжечок при более прогрессирующем течении с GAN. Имеется также умеренная экс-вакуумная дилатация желудочков вследствие прогрессирующей атрофии; C — типичные кудрявые волосы в GAN (т.е. сухие и тугие вьющиеся волосы) часто проявляются у пациентов с рождения или раннего детства; D — у пациентов с GAN развиваются дистальная атрофия и контрактуры, а также может наблюдаться эритема стопы (у детей с GAN часто развивается сколиоз к 8-10 годам)
Гигаксонин экспрессируется в самых разнообразных типах нейрональных клеток и локализуется в аппарате Гольджи и эндоплазматическом ретикулуме. Мутации GAN продемонстрированы в клеточных линиях опухолевых клеток человека, а также в разл. опухолях.
Диагноз м.б. заподозрен клинически на основании раннего начала атаксической походки, признаков невропатии и курчавых или кудрявых волос (см. рис. выше); он подтверждается генетически тестированием гена GAN. Характерны патологические изменения увеличенных или отекших аксонов при биопсии периферических нервов. Клинически появление симптомов происходит в течение первых 5 лет жизни, наблюдается прогрессирующая атаксия и слабость.
По мере прогрессирования заболевания у больных развиваются дисфагия, дизартрия, оптическая нейропатия, ДН, сколиоз (см. рис. выше), а у некоторых на более поздних стадиях возникают приступы. Мутация BAG3, одного из нескольких генов, связанных с миофибриллярной миопатией, также может привести к обнаружению гигантских аксонов гистологически, но клинически отличается от гигантской аксональной невропатии, вызванной мутациями в гене GAN.
з) Гипермиелинизирующая (томакулезная) невропатия; наследственная невропатия с предрасположенностью к компрессионным параличам. Эта наследственная невропатия характеризуется избыточным производством миелина вокруг каждого аксона нерегулярным сегментарным образом, так что в отдельных миелинизированных нервных волокнах возникают томакулезные (колбасообразные) выпуклости. Др. участки того же нерва могут иметь потерю миелина.
Такие нервы особенно подвержены компрессионным параличам, и у пациентов, обычно в подростковом возрасте, наблюдаются рецидивирующие или перемежающиеся мононевропатии, вторичные по отношению к незначительным травмам или компрессионным невропатиям, таким как синдром запястного канала, малоберцовые параличи и даже писчий спазм. Экспрессия фенотипа несколько изменчива. Он передается как АуД-признак, с локусами, идентифицированными на 17p11. 2 и 17р12, и делецией экзонов в гене РМР22 (у некоторых пациентов только микроделеции). Дупликация одного и того же локуса 17р12 приводит к СМТ типа 1А, мутации гена нулевого белка миелина. Биопсия сурального нерва является диагностической, но для наиболее четкой демонстрации аномалий миелина следует сделать специальные препараты с характерными волокнами.
Биопсия кожи или конъюнктивы также м.б. диагностической. Электрофизиол. исследования нервной проводимости нарушены, но неспецифичны. Генетические данные окончательные.
Лечение является поддерживающим и включает предотвращение травм и длительного сдавливания нервов, включая позы сидя или лежа. Иногда показано хирургическая декомпрессия нервов, особенно локтевого нерва.
и) Лейкодистрофии. Некоторые наследственные дегенеративные заболевания белого в-ва ЦНС вызывают периферическую невропатию. Наиболее важными из них являются болезнь Краббе (глобоидная лейкодистрофия), метахроматическая лейкодистрофия и адренолейкодистрофия. В ГМ они производят прогрессирующую, но избирательную демиелинизацию, воздействуя на глубокое белое в-во семиовального центра с относительным сохранением U-волокон вокруг каждой извилины. Дополнительные метаболические нарушения, связанные с периферической невропатией, отмечены в табл. 4.