Синдром прогерии Хатчинсона-Гилфорда, или прогерия, является редким смертельным сегментарным заболеванием с АуД-типом наследования, которое сопровождается преждевременным старением. По оценкам, заболеваемость прогерией при рождении составляет 1:4 млн, а распространенность — 1:20 млн живущих людей; в 2018 г. во всем мире насчитывалось 400 детей, живущих с прогерией. В отношении заболевания нет никаких гендерных, этнических или географических предпосылок.
Прогерия вызвана спорадической АуД-мутацией в гене LMNA, который осуществляет синтез белка ламина А, являющегося основой клеточного ядра и носящего название прогерии. Ламин А — это промежуточный белок внутренней ядерной мембраны, обнаруживаемый в большинстве дифференцированных клеток организма. Без прогерин-специфического лечения у детей с прогерией развивается преждевременный прогрессирующий атеросклероз, и, как правило, они умирают от СН в возрасте от 5 до 20 лет.
Прогерии обнаруживается в повышенной концентрации в коже и сосудистой стенке у здоровых пожилых людей по сравнению с людьми более молодого возраста, что предполагает его роль в нормальном старении.
а) Клинические проявления. У детей развивается внешний вид ускоренного старения, но как клинические, так и биологические совпадения с нормальным старением являются сегментарными или частичными. С возрастом в течение каждого года внешность больных сильно меняется (рис. 1). Ниже описаны клинические проявления по мере их возникновения.
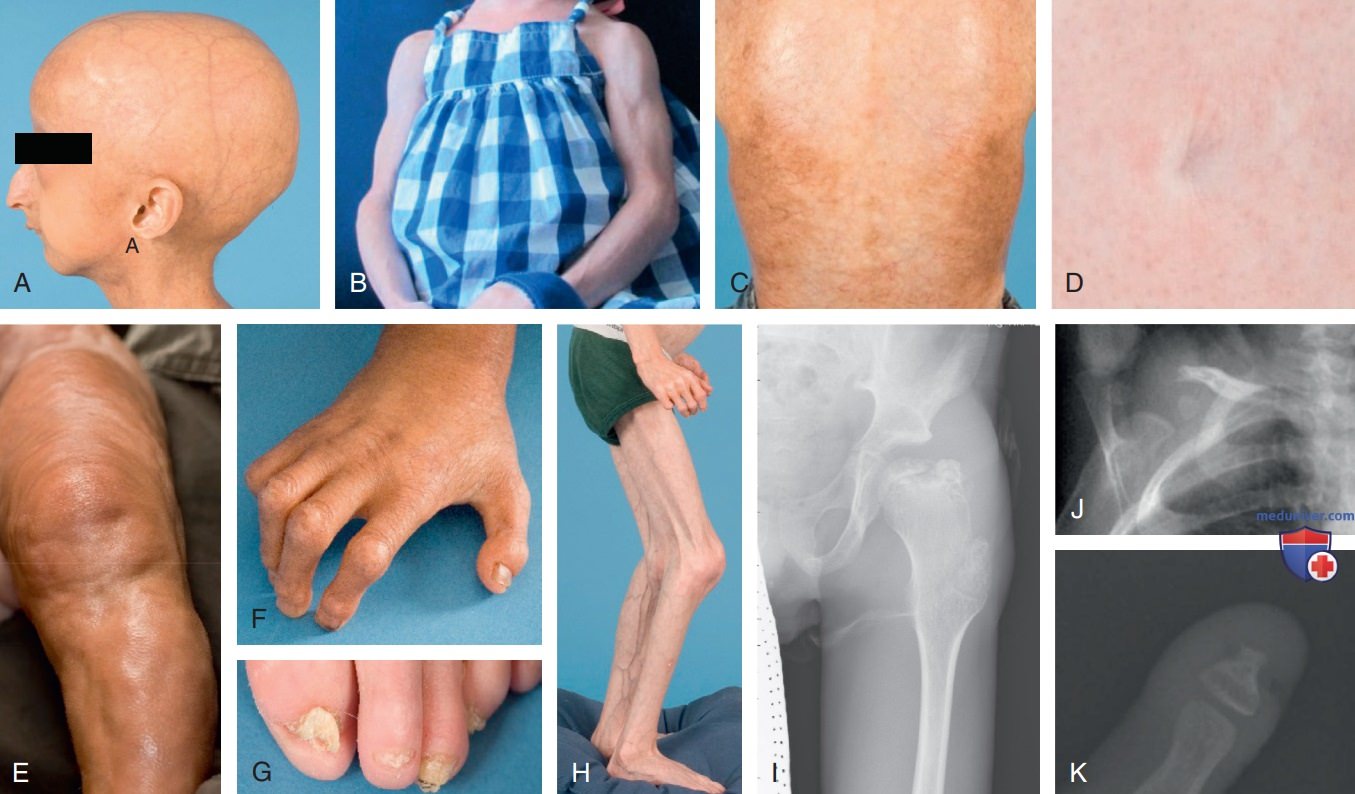
Рисунок 1. Отличительные клинические и рентгенологические признаки синдрома прогерии Хатчинсона-Гилфорда: A — алопеция, выступающие вены кожи головы, узкая переносица, ретрогнатия; B — выступающие контуры мышц за счет генерализованной липоатрофии; C — стянутость и мраморность кожи; D — плоский пупок с рубцом; E — выпячивание кожи; F — контрактуры суставов пальцев кистей; G — дистрофия ногтей, принимающих ложкообразный вид; H — контрактуры коленных суставов, липодистрофия; I — вальгусная деформация шейки бедренной кости (coxa valga); J — остеолизис ключицы; K — акроостеолиз большого пальца кисти
1. Дерматологические изменения. Изменения, которые отмечаются на коже, часто проявляются как первые признаки прогерии. Они различаются по степени тяжести и включают участки обесцвечивания, точечную пигментацию, стянутые участки, которые могут ограничивать движения, а также небольшие (1-2 см) участки туловища или ног, на которых кожа определяется как мягкая и выпуклая. Несмотря на то что больные рождаются с нормальными волосами, волосы на голове теряются в течение первых нескольких лет; после этого на коже головы остаются лишь мягкие, пушистые, редкие и незрелые волосы, при этом отмечается отсутствие бровей и скудное количество ресниц. Дистрофия ногтей возникает в более зрелом возрасте.
2. Отсутствие прибавки массы тела. У детей с прогерией наблюдается нормальное в/утробное и раннее постнатальное развитие. Нарушения в росте и строении тела становятся очевидными в возрасте от нескольких месяцев до 1 года. Это проявляется в виде серьезной задержки физического развития, о чем свидетельствует развитие генерализованной липоатрофии с явным истощением конечностей, периоральный цианоз и выраженная подкожная венозная сеть на голове, шее и туловище.
Средний процентиль МТ обычно нормален при рождении, но снижается до уровня ниже 3-го процентиля, несмотря на адекватное для нормального роста и развития потребление калорий. Обзор 35 детей показал среднее увеличение МТ всего на 0,44 кг в год, начиная с возраста 24 мес, которое сохранялось на протяжении всей жизни. Наблюдаются различия в увеличении МТ между пациентами, однако прогнозируемое увеличение МТ с течением времени у отдельных пациентов является постоянным, линейным и очень предсказуемым; это резко контрастирует с параболической моделью роста у нормальных детей того же возраста и пола.
Средний рост детей с прогерией составляет 1 м, средняя МТ ~15 кг. Показатели окружности головы в норме. Дефицит МТ более выражен, чем дефицит роста, и, поскольку он связан с потерей подкожного жира, приводит к характерному для прогерии истощению. Клинические проблемы, вызванные отсутствием ПЖК, включают чувствительность к холоду и дискомфорт в ногах, вызванный отсутствием амортизации жира. Явный СД при прогерии очень нехарактерен, однако ок. 30-40% детей имеют инсулинорезистентность.
3. Глазные аномалии. Офтальмологические признаки и симптомы частично вызваны стянутостью кожи и недостатком подкожного жира вокруг глаз. Дети часто страдают дальнозоркостью. Вследствие ночного лагофтальма и экспозиционной кератопатии возникают симптомы поражения поверхности глаз, что может привести к изъязвлению и рубцеванию роговицы. Обычным явлением является некоторая степень светобоязни. У большинства пациентов относительно хорошая острота зрения; однако прогрессирование офтальмологического заболевания может привести к снижению остроты зрения. Дети с прогерией должны проходить офтальмологическое обследование при постановке диагноза и далее ежегодно. Рекомендуется достаточное увлажнение поверхности глаза, в т.ч. применение ленточной тарзорафии в ночное время суток.
4. Черепно-лицевые и стоматологические фенотипы. У детей развивается краниофациальная диспропорция с микрогнатией и ретрогнатией по причине гипоплазии нижней челюсти. Типичные в/ротовые и стоматологические проявления включают гиподонтию, замедленное прорезывание зубов, сильную скученность зубов, нёбную дугу, анкилоглоссию, наличие несращения твердого нёба и генерализованную рецессию десен. Прорезывание зубов м.б. отложено на многие месяцы, а молочные зубы могут сохраняться на протяжении всей жизни. Постоянные зубы присутствуют, но могут как прорезываться, так и не прорезываться. Иногда они прорезываются на язычных и небных поверхностях нижнечелюстных и верхнечелюстных альвеолярных гребней, а не на месте первичных резцов. В некоторых, но не во всех случаях удаление молочных зубов способствует перемещению постоянных зубов на место.
5. Нарушения со стороны костной и хрящевой ткани. Развитие костной структуры и плотности костной ткани представляет собой уникальную дисплазию скелета, которая не связана с недостаточностью питания. Акроостеолиз дистальных фаланг и дистальных частей ключиц, тонкие заостренные ребра являются ранними признаками прогерии (уже в возрасте 3 мес). Диспропорция лица, суженная переносица и ретрогнатия чрезвычайно затрудняют интубацию, поэтому рекомендуется использовать волоконно-оптическую интубацию. Грушевидное строение ГК и небольшие ключицы могут привести к развитию нестабильности плечевого сустава. Рост позвоночника и костей таза не нарушен.
Однако диспластический рост оси головки и шейки бедра приводит к вальгусной деформации (т.е. выпрямлению оси головки и шейки бедренной кости >125°) и coxa valga бедра, при котором диаметр головки бедренной кости непропорционально велик для вертлужной впадины, что приводит к нестабильности ТБС.
Возникающая в результате дисплазия ТБС может прогрессировать и приводить к остеоартриту, аваскулярному некрозу, вывиху бедра и неспособности выдерживать МТ. Др. изменения аппендикулярного скелета включают расширение метафизов плечевой и бедренной кости и сужение шейки лучевой кости. Морфология зоны роста в целом остается нормальной, но в пределах одной рентгенограммы может изменяться. Внешний вид очагов окостенения, используемых для определения костного возраста, является нормальным. Костная структура, оцененная с помощью периферической количественной КТ лучевой кости, демонстрирует отчетливые и серьезные отклонения в геометрии структуры кости, что характерно для прогерии, представляющей скелетную дисплазию.
Минеральная плотность костной ткани (aBMD; англ. areal bone mineral density) z, измеренная с помощью двухэнергетической рентгеновской абсорбциометрии (DXA; англ. Dual-energy X-ray absorptiometry) с поправкой на рост и возраст, и истинная (объемная) минеральная плотность костной ткани, оцененная с помощью периферической количественной КТ, показывают нормальные или умеренно сниженные значения, что опровергает предположение о том, что пациенты с прогерией имеют остеопороз.
Частота переломов при прогерии нормальная и не связана с патологическими переломами, наблюдаемыми при др. метаболических заболеваниях костей среди детей, таких как несовершенный остеогенез.
Контрактуры некоторых суставов (напр., суставов пальцев, локтевых, ТБС, коленных, голеностопных) могут присутствовать при рождении и прогрессировать с возрастом из-за слабости тканевых структур, окружающих сустав (суставной капсулы, связок, кожи). Наряду с нарушением конгруэнтности суставных поверхностей эти изменения ограничивают объем движений в суставах и отражаются на походке. Рекомендуются регулярные занятия лечебной физкультурой на протяжении всей жизни с целью максимального увеличения функции суставов.
6. Слух. Развитие низкочастотной кондуктивной потери слуха широко распространено при прогерии и свидетельствует о ригидности барабанной перепонки и/или дефиците костных структур и связочного аппарата среднего уха. В целом это не влияет на способность слышать обычную разговорную речь, но рекомендуется размещение на первых рядах в классе и проведение ежегодных проверок слуха.
7. Сердечно-сосудистые заболевания. Примерно 80% случаев смерти от прогерии вызваны СН, которая может развиваться вследствие перенесенной респираторной суперинфекции или на фоне проведения хирургического вмешательства. Прогерия — это первичная васкулопатия, характеризующаяся повсеместным ускоренным сужением просветов сосудов, следствием которых является окклюзионное поражение крупных и мелких сосудов из-за образования атеросклеротических бляшек и развитие клапанной и СН в более поздние годы. ГБ, стенокардия, кардиомегалия, метаболический синдром и ХСН являются частыми явлениями терминальной стадии.
В ходе исследования по изучению трансторакальной ЭхоКГ у пациентов, ранее не получавших лечения, выявили диастолическую дисфункцию ЛЖ связанную с возрастным снижением показателей боковой и септальной ранней (Е’) диастолической тканевой доплеровской скорости z и увеличением отношения митрального притока (Е) к показателям боковой и септальной скорости z. Др. результаты ЭхоКГ включали гипертрофию ЛЖ, систолическую дисфункцию ЛЖ и поражение митрального или аортального клапана. Они, как правило, появляются в более позднем возрасте. Рекомендуется рутинное УЗИ сонной артерии для мониторинга образования бляшек и определения скорости распространения пульсовой волны каротиднофеморальным методом (PWVcf; англ. carotid femoral pulse wave velocity) для определения жесткости сосудов и ЭхоКГ.
8. Церебральная артериопатия и инсульт. Инфаркт ГМ может возникнуть, когда у ребенка нормальная ЭКГ. Самый ранний инсульт случился у ребенка в возрасте 0,4 года. Чаще инсульты случаются в более позднем возрасте. На протяжении всей жизни МРТ-свидетельства инфаркта могут быть обнаружены у 60% пациентов с прогерией, причем клинически половина из них не проявляется. Обнаруживаются заболевания как крупных, так и мелких сосудов; происходит обширное образование коллатеральных сосудов. Закупорки сонной артерии хорошо описаны, однако инфаркт может возникнуть даже при их отсутствии. Склонность к инсультам и лежащая в их основе жесткая сосудистая сеть делают поддержание адекватного АД за счет гидратации (привычное хорошее питье) приоритетом у пациентов с прогерией; следует проявлять особую осторожность при рассмотрении вопроса о поддержании постоянного АД во время общей анестезии, авиаперелетов и жаркой погоды.
Кроме того, 15% случаев летальных исходов среди детей с прогерией происходят от повреждений или травм головы, включая субдуральную гематому. Это указывает на предрасположенность таких детей к развитию субдуральных гематом.
9. Сексуальное развитие. У женщин с прогерией могут развиться вторичные половые признаки II стадии по шкале Таннера, включая признаки раннего развития МЖ и появление редких волос на лобке. Они не достигают стадии Таннера III. Несмотря на минимальные или отсутствующие физические признаки полового созревания и минимальное количество жира в организме, более чем у половины девушек менархе наступает спонтанно в среднем в возрасте 14 лет. У девушек с наступившим менархе, по сравнению с женщинами без менструации, показатели МТ, процентное содержание жира в организме и уровни лептина в сыворотке крови схожи, но значительно ниже, чем у здоровых подростков. Если кровотечение становится сильным, показатели ОАК могут быть снижены и для уменьшения тяжести кровотечения можно использовать оральные контрацептивы. Вторичные половые признаки у лиц мужского пола не изучались. Задокументированных случаев репродуктивной способности у женщин или мужчин с прогерией нет.
10. Нормально функционирующие системы. Функция печени, почек, ЩЖ, иммунной, пищеварительной и НС (за исключением случаев, связанных с инсультом) не изменяются. Уровень интеллекта сохраняется нормальным для возраста, возможно, отчасти из-за подавления экспрессии прогерина в ГМ специфической для ГМ микро-РНК, miRNA-9.
б) Лабораторные признаки. Наиболее достоверными лабораторными данными являются уровень лептина в сыворотке ниже определяемого уровня (>90%) и резистентность к инсулину (60%). Уровень содержания тромбоцитов зачастую бывает умеренно высоким. Концентрации адипонектина и ЛПВП снижаются с возрастом до уровней значительно ниже нормы. В остальном липидные панели, высокочувствительный СРБ, биохимический анализ крови, результаты функциональных тестов состояния печени и почек, эндокринных тестов и тестов на коагуляцию в целом имеют нормальные значения.
в) Молекулярный патогенез. Причиной развития прогерии являются мутации в гене LMNA. Нормальный ген LMNA/C кодирует белки ламин А и С, из которых только ламин А связан с заболеваниями человека. Белки ламина — это основные белки ядерной ламины, сети из белков промежуточных филаментов сложного молекулярного интерфейса, расположенного между внутренней мембраной ядерной оболочки и хроматином. Целостность этой ламины занимает центральное место для многих клеточных функций, создания и поддержания структурной целостности ядерного каркаса, репликации ДНК, транскрипции РНК, организации ядра, сборки ядер-ных пор, функции хроматина, циклирования клеток, старения и апоптоза.
Прогерия практически всегда является спорадическим АуД-заболеванием. Есть два описанных случая родства между сиблингами, оба предположительно происходящие из родительского мозаицизма, когда у одного фенотипически нормального родителя присутствует мозаицизм зародышевой линии. Это вызвано ускоренным использованием альтернативного внутреннего сайта соединения, что приводит к удалению 150 пар оснований в 3’-части экзона 11 гена LMNA. Примерно в 90% случаев это происходит из-за одиночной замены цитозина (С) на тимин (Т) в нуклеотиде 1824, о котором не упоминается (Gly608Gly), но который оптимизирует место внутреннего сплайсинга в экзоне 11. В остальных 10% случаев наблюдается одна из нескольких одноосновных мутаций в донорном месте сплайсинга интрона 11, что снижает специфичность данного места и изменяет баланс сплайсинга в пользу внутреннего сплайсинга.
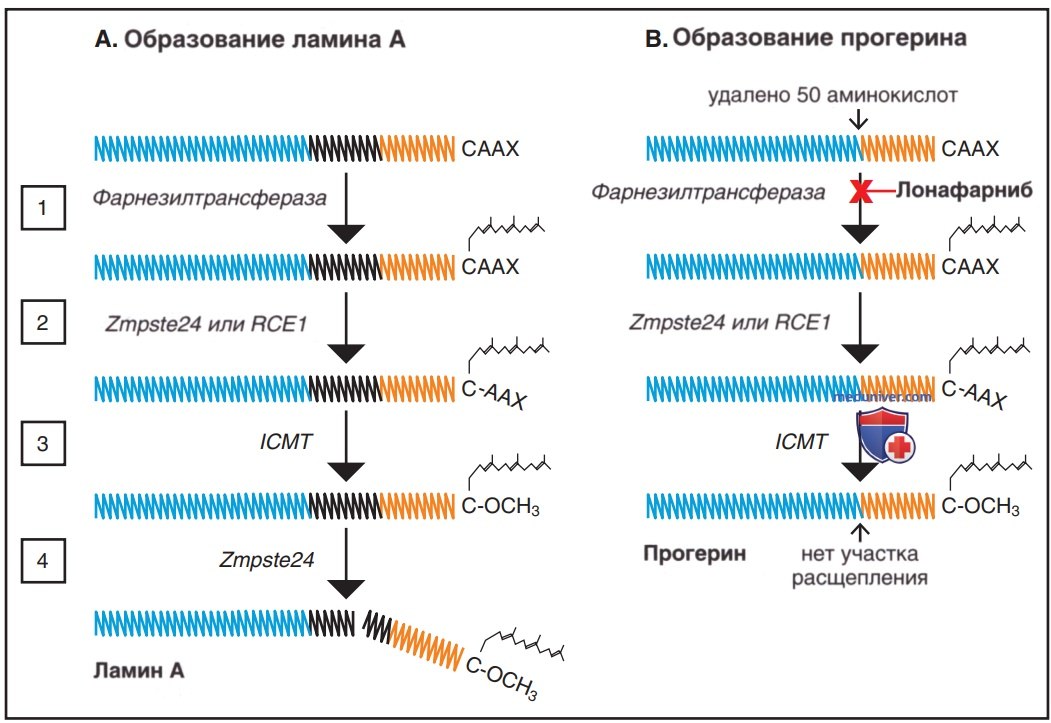
Вслед за всеми этими мутациями трансляция с последующим посттрансляционным процессингом измененной мРНК продуцирует прогерии, укороченный аномальный белок ламина А с 50-аминокислотной делецией около его С-конца. Понимание посттрансляционного процесса обработки и того, как он изменяется с образованием прогерина, привело к получению ряда перспектив лечения этого заболевания (рис. 2).
Рисунок 2. Пути посттрансляционной обработки, продуцирующие ламин А и прогерии, включая целевой участок для лонафарниба. А — полипептидная цепь Prelamin А, показывающая ее центральный домен а-спирального стержня и карбоксильный конец молекулы — СААХ (С — цистеин, АА — две алифатические аминокислоты, X — любая аминокислота). Домен а-спирального стержня разделен на сегменты, которые способствуют отображению дефекта прогерина. Посттрансляционный процессинг состоит из четырех этапов: 1 — фарнезильная группа присоединяется к цистеиновому остатку СААХ-бокса с помощью фарнезилтрансферазы; 2 — последние три остатка протеолитически расщепляются металлопротеазой цинка Zmpste24 или ферментом, превращающим Ras (RCE1); 3 — карбоксиметилирование изопренилцистеинкарбоксилметилтрансферазой (ICMT); 4— 15 С-концевых остатков, включая фарнезилированный и карбоксиметилированный цистеин, отщепляются Zmpste24. B — А 50-аминокислотная делеция в преламине А (представленная черным сегментом стержня ламина А) является результатом мутации, которая активирует криптический участок сплайсинга в экзоне 11-го гена LMNA. Эта делеция оставляет прогерии без места присоединения для последней стадии процессинга — отщепления фарнезилированных и карбоксиметилированных концевых 15 аминокислотных остатков. Т.о., прогерин остается фарнезилированным и встроенным во внутреннюю ядерную мембрану, где и вызывает значительную часть клеточного повреждения
И ламин А, и прогерии обладают метилированной фарнезильной боковой группой, присоединенной во время посттрансляционного процессинга. Это липофильный фрагмент, который облегчает включение белков во внутреннюю ядерную мембрану, где выполняется большая часть функций ламина и прогерина. Для формирования нормального ламина А потеря метилированной фарнезильной «точки привязки» высвобождает преламин из ядерной мембраны, делая его растворимым для аутофагической деградации. Однако прогерии сохраняет свою фарнезильную часть. Он остается прикрепленным к мембране, связывая др. белки и вызывая образование пузырей в ядре, нарушая митоз и изменяя экспрессию генов. Прогерии также сохраняет метильную часть.
Развитие заболевания при прогерии связано с доминантным отрицательным механизмом. Фенотип данного заболевания обусловлен скорее воздействием прогерина, а не снижением уровней ламина А. Тяжесть заболевания определяется отчасти уровнями прогерина, которые зависят от наличия определенной мутации, типа ткани или др. факторов, влияющих на использование участка внутреннего сплайсинга.
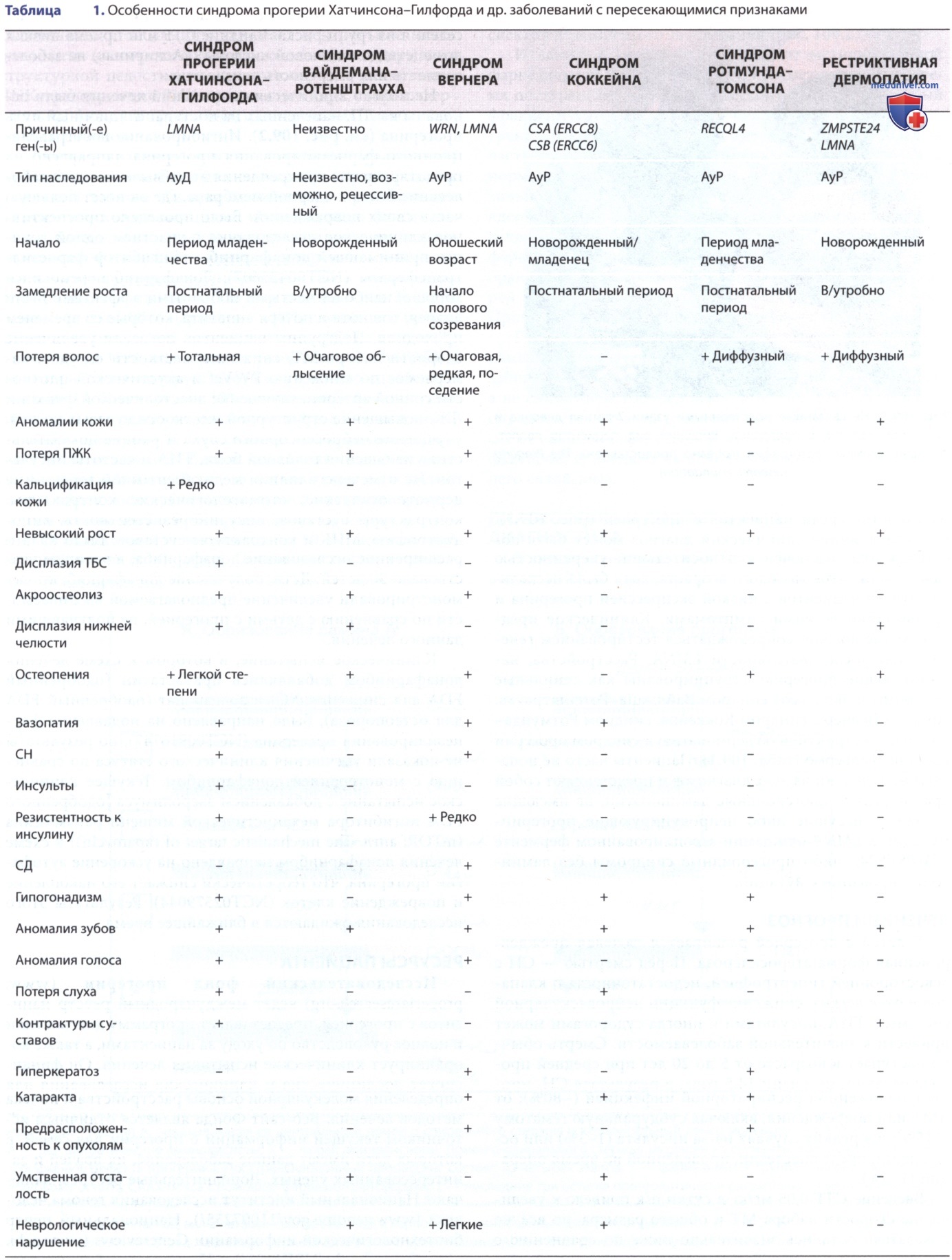
г) Диагностика и дифференциальная диагностика. В целом сочетание небольшого размера тела, костей, волос, ПЖК и кожи приводит к заметному физическому сходству среди пациентов с прогерией (рис. 3). По этой причине клинический диагноз может быть поставлен или исключен с относительной уверенностью даже у пациентов молодого возраста, хотя было несколько случаев пациентов с низкой экспрессией прогерина и чрезвычайно легкими симптомами. Клиническое предположение должно сопровождаться тестированием генетической последовательности LMNA. Расстройства, напоминающие прогерию, сгруппированы как сенильные синдромы и включают синдром Вайдмана-Ротенштрауха, синдром Вернера, синдром Коккейна, синдром Ротмунда-Томсона, рестриктивную дермопатию и синдром прогерии Нестора-Гильермо (табл. 1). Пациенты часто не попадают ни под один из этих диагнозов и представляют собой крайне редкие прогероидные ламинопатии, не имеющие названия, несущие либо непродуцирующие прогерин-мутации в LMNA или ламин-ассоциированном ферменте (ZMPSTE24), либо прогероидные синдромы без ламин-ассоциированных мутаций.
Рисунок 3. Не связанные родственными узами 7-летняя девочка и 10-летний мальчик с прогерией. Внешний вид пациентов является очень схожим
д) Лечение и прогноз. У детей с прогерией развивается тяжелая преждевременная форма атеросклероза. Перед смертью — СН с левосторонней гипертрофией, недостаточностью клапанов и отек легких; снижение функции нейроваскулярной системы с ТИА, инсультами и иногда судорогами может привести к значительной заболеваемости. Смерть обычно наступает в возрасте от 5 до 20 лет при средней продолжительности жизни 14,5 года, в результате СН, иногда с наложенной респираторной инфекцией (80%); от ЧМТ или повреждения, включая субдуральную гематому (15%); и в редких случаях из-за инсульта (1-3%) или осложнений после анестезии, проведенной во время операции (1-3%).
Введение СТГ 0,05 мг/кг в сутки п/к привело к увеличению скорости набора МТ и общего размера, но все же показатели остались значительно ниже по сравнению с нормальными детьми. Терапия ацетилсалициловой кислотой («Аспирином») в низких дозах рекомендуется в дозе 2 мг/кг в сутки, в дополнение к тому, что известно о снижении сердечно-сосудистой патологии у взрослого населения из групп риска. Влияние СТГ или приема низких доз ацетилсалициловой кислоты («Аспирина») на заболеваемость или смертность неизвестно.
Несколько клинических испытаний лечения были основаны на ЛП, нацеленных на посттрансляционный путь прогерина (см. рис. 2). Ингибирование посттрансляционного фарнезилирования прогерина направлено на предотвращение прикрепления этого вызывающего заболевание белка к ядерной мембране, где он несет большую часть своих повреждений. Было проведено проспективное клиническое исследование с участием одной группы, принимавшей лонафарниб — ингибитор фарнезил-трансферазы (NCT00425607). Лонафарниб переносился хорошо; наиболее частыми побочными эффектами были диарея, тошнота и потеря аппетита, которые со временем проходили.
Подгруппы пациентов показали увеличение скорости набора МТ, снижение жесткости сосудов, измеряемое по снижению PWVcf и акустической плотности сонной артерии, улучшение диастолической функции ЛЖ, повышение структурной жесткости лучевых костей, улучшение нейросенсорного слуха и ранние доказательства уменьшения головной боли, ТИА и частоты инсультов. Не отмечено влияния медикаментозной терапии на дерматологические, стоматологические контрактуры, контрактуры суставов, инсулинорезистентность, липодистрофию, МПК и контрактуры суставов.
Было начато расширенное исследование лонафарниба, в котором участвовало 30 детей. Дети, получавшие лонафарниб, продемонстрировали увеличение предполагаемой выживаемости по сравнению с детьми с прогерией, не получавшими данного лечения.
Клиническое испытание, в котором к схеме лечения лонафарнибом добавлялись правастатин (одобренный FDA для снижения ХС) и золедронат (одобренный FDA для остеопороза), было направлено на подавление фарнезилирования прогерина (NCT00916747), но результаты не показали улучшения клинического статуса по сравнению с монотерапией лонафарнибом. Текущее клиническое испытание с добавлением эверолимуса [одобренного FDA ингибитора механистической мишени рапамицина (mTOR; англ. The mechanistic target of rapamycin)] к схеме лечения лонафарнибом направлено на ускорение аутофагии прогерина, что теоретически снижает его накопление и повреждение клеток (NCT02579044). Результаты этого исследования ожидаются в ближайшее время.
д) Ресурсы пациента. Исследовательский фонд прогерии ведет международный реестр пациентов с прогерией, предоставляет программу диагностики и полное руководство по уходу за пациентами, а также координирует клинические испытания лечения. Он финансирует доклинические и клинические исследования для определения молекулярной основы расстройства и поиска методов лечения. Веб-сайт Фонда является отличным источником текущей информации о прогерии для семей, в которых дети имеют данное заболевание, их врачей и заинтересованных ученых. Дополнительные ресурсы включают сайт МедУнивер, Национальный институт исследования генома человека, Национальный центр биотехнологической информации Genereviews и Национальный центр развития трансляционных наук.