Порфирия, связанная с дефектом дегидратазы δ-аминолевулиновой кислоты (АДП), иногда называется порфирией Досса в честь имени исследователя, описавшего первые случаи заболевания. Термин «плюмбопорфирия» подчеркивает сходство этого состояния с отравлением свинцом, но неправильно подразумевает, что оно вызвано воздействием свинца.
а) Этиология. Эта порфирия возникает из-за дефицита ALAD. Тип наследования — АуР. При анализе мутаций гена было подтверждено только шесть случаев. Распространенность гетерозиготной недостаточности ALAD оценивается в <1% в Германии и ~2% в Швеции.
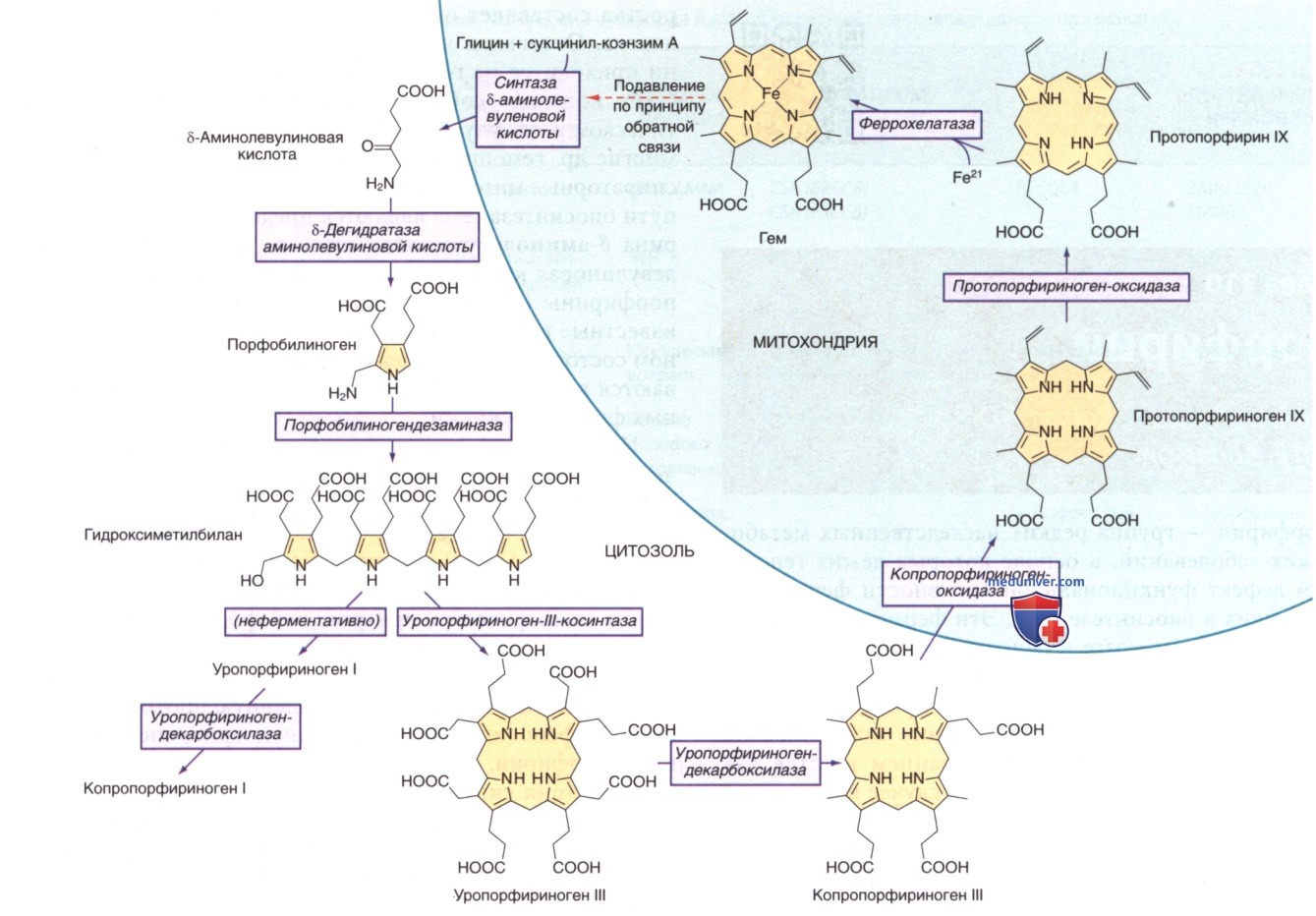
б) Патология и патогенез. ALAD катализирует конденсацию двух молекул АЛК с образованием пиррольного ПБГ (см. рис. ниже). Фермент ингибируется рядом экзогенных и эндогенных хим. в-в. ALAD является основным связывающим свинец белком в эритроцитах, и свинец может замещать атомы цинка в ферменте. Подавление активности ALAD-эритроцитов является чувствительным показателем воздействия свинца.
Ферменты и промежуточные продукты пути биосинтеза гема. Этот путь регулируется в печени конечным продуктом, гемом, в основном за счет подавления обратной связи (пунктирная стрелка)
Было идентифицировано 11 аномальных аллелей ALAD, большинство из которых имеют точечные мутации; некоторые из них проявляют частичную активность, так что синтез гема частично сохраняется. Количество остаточной активности фермента может предопределить фенотипическую тяжесть этого заболевания.
АДП часто классифицируют как печеночную порфирию, хотя место гиперпродукции АЛК не установлено. Пациенту, имеющему тяжелое заболевание, на ранней стадии была проведена трансплантация печени без значительного клинического или биохимического улучшения, что может свидетельствовать о том, что в печени не возникало избытка промежуточных продуктов.
Избыток копропорфирина III в моче при АДП, вероятно, может возникать в результате метаболизма АЛК до порфириногенов в тканях, отличных от тканей с избыточной продукцией АЛК. Введение больших доз АЛК нормальным людям также приводит к развитию значительной копропорфиринурии.
Повышенный уровень протопорфирина в эритроцитах, как и при всех др. гомозиготных порфириях, можно объяснить накоплением промежуточных продуктов более раннего пути метаболизма в эритроидных клетках костного мозга во время синтеза Hb с последующим их преобразованием в протопорфирин после завершения синтеза Hb. Неврологические симптомы объясняются нейротоксическим действием АЛК, но это не является доказанным.
в) Клинические проявления. В большинстве случаев симптомы напоминают др. острые порфирии, включая острые приступы боли в животе и периферическую невропатию. Действие провоцирующих факторов, таких как воздействие вредных ЛП, в большинстве случаев не выражено.
В четырех случаях из всех зарегистрированных нарушения наблюдались у подростков. У шведского младенца было более тяжелое заболевание с неврологическими нарушениями и задержкой развития. У 63-летнего мужчины из Бельгии развилась острая моторная полиневропатия одновременно с миелопролиферативным заболеванием.
г) Лабораторные признаки. Повышение экскреции АЛК с мочой; копропорфирин III и цинк-протопорфирин эритроцитов значительно повышены. Экскреция ПБГ с мочой в норме либо повышена незначительно. Активность ALAD-эритроцитов заметно снижена, у родителей больных она составляла ~50% от нормы, также у родителей сохранялся нормальный уровень содержания АЛК в моче.
д) Диагностика и дифференциальная диагностика. Остальные три острые порфирии характеризуются значительным увеличением как АЛК, так и ПБГ. Напротив, АЛК (но не ПБГ) существенно ↑ при АДП. Значительный дефицит ALAD-эритроцитов и 50-процентная активность данного фермента у родителей больного служат подтверждением диагноза. Необходимо исключить др. причины дефицита ALAD, такие как отравление свинцом.
Сукцинилацетон накапливается при наследственной тирозинемии 1-го типа и структурно подобен АЛК, ингибирует ALAD и может вызывать повышенную экскрецию АЛК с мочой и клинические проявления, напоминающие острую порфирию. Сообщалось о приобретенном идиопатическом дефиците ALAD. В отличие от отравления свинцом, недостаточная активность ALAD при АДП не восстанавливается при добавлении in vitro сульфгидрильных реагентов, таких как дитиотреитол.
Даже если не обнаружены др. причины дефицита ALAD, важно подтвердить диагноз АДП молекулярными генетическими исследованиями.
е) Лечение. Опыт лечения АДП ограничен, но аналогичен лечению др. острых порфирий. По всей видимости, глюкоза имеет минимальную эффективность, но можно попробовать ее назначить при легких симптомах. Терапия с использованием гемина оказалась эффективной при острых приступах у подростков мужского пола, а еженедельные инфузии предотвратили приступы у двух из этих пациентов.
Гемин не показал свою эффективность ни в биохимическом, ни в клиническом отношении при его назначении шведскому ребенку с тяжелой формой заболевания; он вызывал биохимический ответ, но не давал клинического улучшения у бельгийского мужчины с поздней формой развития болезни, у которого наблюдалась периферическая невропатия, но не было острых приступов.
Гемин также эффективен при лечении симптомов, подобных порфирии, связанных с наследственной тирозинемией, он может значительно ↓ уровень АЛК и копропорфирина в моче при отравлении свинцом. Рекомендуется избегать ЛП, которые вредны при др. острых порфириях. Трансплантация печени оказалась неэффективной у ребенка с тяжелым заболеванием.
ж) Прогноз. В типичных случаях АДП прогноз в целом является благоприятным, несмотря на то что могут возникать рецидивирующие приступы. Течение было неблагоприятным у шведского ребенка (с более тяжелым заболеванием) и неопределенным у взрослых с поздним началом заболевания (связанного с миелопролиферативными нарушениями).
з) Профилактика и генетическое консультирование. Гетерозиготные родители должны знать, что их последующие дети подвержены риску заболевания АДП, равно как и любым др. АуР-заболеванием. Возможна постановка диагноза в пренатальном периоде, но на практике о таких случаях не сообщалось.