Поздняя кожная порфирия (ПКП) — наиболее распространенная и легко поддающаяся лечению порфирия, которая встречается у человека (см. табл. 2). Она возникает в среднем или пожилом возрасте и редко встречается у детей. Предыдущие термины включают симптоматическую порфирию, симптоматическую ПКП и идиосинкразическую порфирию. Основная причина — специфическая для печени приобретенная недостаточность уропорфириногендекарбоксилазы (УПГД) с участием нескольких типов генетических и приобретенных факторов восприимчивости, включая гетерозиготные мутации УПГД при семейном ПКП.
Гепатоэритропоэтическая порфирия (ГЭП) — гомозиготная форма семейного ПКП — обычно имеет более тяжелую форму в детстве, клинически напоминающую ВЭП.
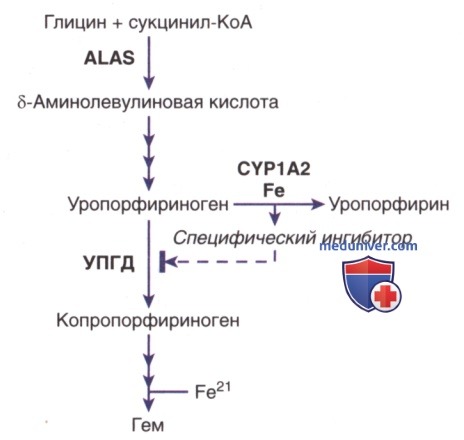
а) Этиология. ПКП вызывается снижением активности УПГД в печени до <20% от нормальной активности. Ингибитор печеночного УПГД был охарактеризован как уропорфометен, который образуется в результате частичного окисления ферментного субстрата уропорфириногена.
В процессе его синтеза участвуют цитохромы, напр. CYP1A2, а также железо (рис. 1). Несмотря на подавление активности фермента, количественное содержание белка печеночного фермента, по данным иммуногистохим. анализа, остается на уровне, предопределенном генетически.
Рисунок 1. Синтез специфического ингибитора уропорфириногендекарбоксилазы в печени при поздней кожной порфирии. ALAS — δ-аминолевулиновой кислоты синтаза; CYP1A2 — цитохром Р450 1А2; УПГД — уропорфириногендекарбоксилаза
УПГД катализирует декарбоксилирование четырех боковых цепей уксусной кислоты уропорфириногена (октакарбоксилпорфириноген) и формирование копропорфириногена (тетракарбоксилпорфириноген). Ферментная реакция проходит последовательно, а именно по часовой стрелке с образованием промежуточных продуктов — гепта-, гекса- и пентакарбоксилпорфириногенов. Уропорфириноген III является более предпочтительным субстратом, чем др. уропорфириногены. УПГД человека представляет собой димер с двумя активными центрами, щели которых располагаются бок о бок.
Ген УПГД локализуется на хромосоме 1p34. Он содержит 10 экзонов и всего один промотор. По этой причине указанный ген во всех тканях транскрибируется в виде единственной мРНК.
У основной части пациентов с ПКП (80%) мутации в гене УПГД отсутствуют и заболевание носит спорадический характер (тип 1). Некоторые пациенты являются гетерозиготными носителями мутантного гена УПГД и страдают семейной формой (тип 2) ПКП. Описаны такие виды мутаций, как миссенс-мутации, нонсенс-мутации и мутации сайтов сплайсинга, несколько малых и больших делеций, малые инсерции, причем лишь некоторые из них идентифицированы более чем в одном семействе.
Некоторые из этих мутаций м.б. локализованы вблизи щели активного центра, но большинство, по всей видимости, затрагивают области, играющие важную структурную роль. Гетерозиготности по мутации УПГД недостаточно, чтобы вызвать ПКП. У лиц с ПКП 2-го типа с рождения процент активности УПДГ составляет 50%. В более позднем возрасте др. провоцирующие факторы (как в случае с типом 1) приводят к выработке ингибитора уропорфометена и дальнейшему снижению активности УПДГ в печени до <20% от нормы. Поскольку пенетрантность генетического признака низкая, многие пациенты с семейным ПКП не имеют семейного анамнеза болезни.
Индукция печеночного ALAS1 не является важной особенностью при ПКП, хотя алкоголь может немного ↑ содержание этого фермента. Железо и эстрогены не являются мощными индукторами ALAS1, а ЛП, которые являются мощными индукторами ALAS1 и ферментов цитохрома P450, гораздо реже вовлекаются при ПКП, чем при лечении острой порфирии.
Волдыри на коже возникают в результате выделения порфиринов из печени. Воздействие солнечного света приводит к образованию активных форм кислорода в коже, активации комплемента и повреждению лизосом.
б) Эпидемиология. Различия в распространенности, вероятно, связаны с географическими различиями в отношении провоцирующих факторов, таких как гепатит С и употребление этанола. Ежегодная заболеваемость в Соединенном Королевстве оценивалась в 2-5:100 0000 населения в целом, а распространенность в США и Чехословакии — как 1:25 000 и 1:5000 среди всего населения соответственно. Сообщалось, что заболевание было распространено среди народности банту в Южной Африке в связи с гемосидерозом. ПКП чаще встречается у мужчин, возможно, из-за повышенного потребления алкоголя, а у женщин заболевание обычно связано с приемом эстрогенов.
В 1950-х гг. массовая вспышка ПКП произошла в восточной части Турции. Пшеница, предназначенная для посадки и обработанная гексахлорбензолом в качестве фунгицида, многими употреблялась в пищу во время нехватки продовольствия. Сообщалось о случаях и небольших вспышках ПКП после воздействия др. хим. в-в, включая ди- и трихлорфенолы и 2,3,7,8-тетрахлордибензо-n-диоксин (ТХДД, диоксин). В большинстве случаев проявления ↓ после прекращения воздействия. Зарегистрированы случаи отсроченного начала — через несколько лет после воздействия хим. в-в.
в) Патология и патогенез. ПКП подразделяется на три клинически похожих типа. Генерация ингибитора УПДГ в печени играет важную роль в каждом из трех типов. У 80% пациентов с типом 1 (спорадическим) ПКП нет мутаций UROS, а активность УПДГ является нормальной во внепеченочных тканях, таких как эритроциты. При типе 2 ПКП (семейном) гетерозиготная мутация UROS приводит к частичному (50%) дефициту УПДГ во всех тканях с рождения, и у некоторых гетерозигот при дальнейшем снижении активности УПДГ в печени до <20% от нормы заболевание переходит в активную форму.
ГЭП возникает в результате наследования мутации UROS от каждого родителя и обычно вызывает серьезную фоточувствительность, напоминающую ВЭП, начиная с раннего детства. У некоторых компаунд-гетерозигот в детстве развились симптомы, более типичные для ПКП. Тип 3 встречается редко и описывает ПКП без мутации UROS, встречающейся у >1 члена семьи. Др. генетическая основа, такая как мутации HFE, м.б. идентифицирована у пациентов с типом 3.
Цитохромы, особенно CYP1A2, могут катализировать окисление уропорфириногена до уропорфирина. Такая активность уропорфириногеноксидазы усиливается железом и приводит к образованию ингибитора УПДГ (рис. 1). CYP1a2 кажется важным для развития уропорфирии у грызунов, поскольку экспериментальная уропорфирия не развивается у мышей с нокаутом гена CYPla2.
г) Провоцирующие факторы. У пациентов с ПКП к провоцирующим факторам и в разл. сочетаниях могут относиться следующие.
• Железо. Нормальное или повышенное количество железа в печени имеет большое значение для развития ПКП, и лечение кровопусканием для снижения уровня железа в печени приводит к ремиссии. Уровни ферритина в сыворотке обычно находятся на верхней границе нормального диапазона или умеренно повышены, а гистология печени нередко показывает повышенное содержание железа. Распространенность мутации C282Y гена HFE (гена гемохроматоза), которая является основной причиной гемохроматоза у людей североевропейского происхождения, ↑ при ПКП как 1-го, так и 2-го типа; 10% пациентов являются гомозиготами по C282Y.
В южной части Европы более распространена мутация H63D.
ПКП может развиться у пациентов с вторичной перегрузкой железом. Сниженная экспрессия гепсидина в печени происходит при гемохроматозе, а также при ПКП, независимо от генотипа HFE, что может объяснять развитие сидероза печени при этом состоянии.
• Гепатит С. Вирусный гепатит С широко распространен при ПКП в большинстве географических регионов; в США, напр., вирусный гепатит С присутствует в 56-74% случаев, что аналогично показателям в Южной Европе.
Распространенность гепатита С при ПКП ниже в Северной Европе (<20%). Стеатоз и окислительный стресс при HBV могут способствовать опосредованной железом генерации активных форм кислорода и ингибитора УПДГ. Регуляция гепсидина нарушается при гепатите С, что может приводить к повышенной абсорбции железа.
• Вирус иммунодефицита человека. Множество сообщений указывают на то, что ВИЧ-инфекция может способствовать развитию ПКП, хотя и реже, чем HCV.
• Этанол. Давно признанная связь между алкоголем и ПКП м.б. объяснена генерацией активной формы кислорода, которая может вызывать окислительное повреждение, повреждение митохондрий, истощение восстановленного глутатиона и др. антиоксидантных защитных механизмов, повышение выработки эндотоксина и активацию клеток Купфера. Кроме того, алкоголь может нарушать синтез гепсидина, способствуя тем самым перегрузке железом.
• Курение и цитохромы Р450. Курение как провоцирующий фактор широко не изучалось, но оно часто связано с употреблением алкоголя при ПКП. Курение может привести к индукции цитохромов печени и окислительному стрессу. Считается, что ферменты цитохрома Р450 печени играют важную роль в окислении уропорфириногена и создании ингибитора УПДГ (см. рис. 1). Генетический полиморфизм CYP1A2 и CYP1A1 вовлечен в развитие ПКП у человека. Индуцибельный генотип CYP1A2 был более распространен среди пациентов с ПКП, чем в контрольной группе, в нескольких исследованиях.
• Антиоксидантный статус. Дефицит аскорбиновой кислоты способствует развитию уропорфирии на лабораторных моделях и, возможно, при ПКП у человека. В одной серии исследований уровни аскорбиновой кислоты в плазме были существенно снижены у 84% пациентов с ПКП. Были описаны низкие уровни каротиноидов в сыворотке, что также свидетельствует о важности окислительного стресса в гепатоцитах при ПКП.
• Эстрогены. Использование эстроген-содержащих оральных контрацептивов или заместительной эстрогеновой терапии в постменопаузе часто связано с развитием ПКП (типа 1 или 2) у женщин. Иногда ПКП возникает во время беременности, при этом остается неясным, увеличивается ли при этом риск развития заболевания.
е) Клинические проявления:
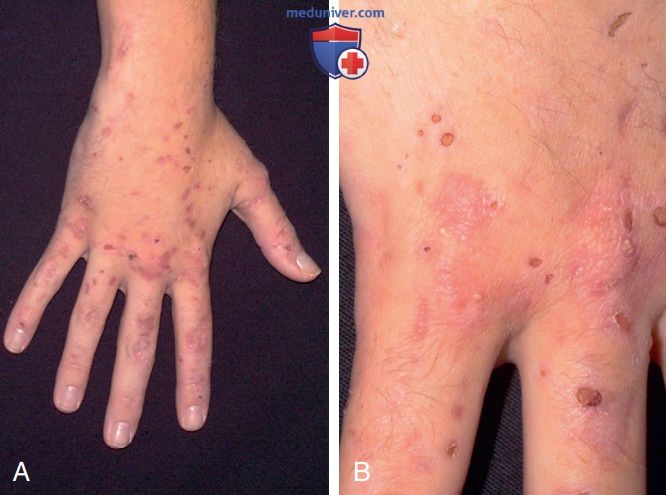
1. Кожные проявления. ПКП легко распознается по появлению волдырей и струпьев на коже на тыльной стороне рук, которые являются наиболее подверженными солнечному воздействию участками тела; несколько реже они появляются на предплечьях, лице, ушах, шее, ногах и ступнях (рис. 2). Пузыри, заполненные жидкостью, обычно разрываются и покрываются струпьями или в виде эрозий, которые медленно заживают и инфицируются. Кожа на тыльной стороне рук обычно мягкая, причем при незначительной травме могут появиться волдыри или эрозии кожи.
Рисунок 2. Поздняя кожная порфирия (ПКП): А — правая рука пациента с ПКП с многочисленными эрозиями и эритематозными пятнами; В — крупный план правой руки
Маленькие белые папулы, называемые милиумами, могут предшествовать или следовать за образованием пузырей на коже. Также распространены гипертрихоз и гиперпигментация кожи лица. Сильное рубцевание и утолщение кожи, подвергшейся солнечному воздействию, может напоминать склеродермию. Результаты биопсии кожи включают субэпидермальные пузыри и отложение шифф «+» в-в* вокруг кровеносных сосудов и тонкого фибрилл в дермоэпителиальном слое, что м.б. связано с чрезмерной хрупкостью кожи. IgG, др. Ig и комплемент также откладываются в дермоэпителиальном слое и вокруг кровеносных сосудов дермы.
Поражения кожи и изменения на гистологическом уровне не специфичны для ПКП. Такие же результаты наблюдаются при ВП и НКП и напоминают результаты ВЭП и ГЭП, но обычно они менее серьезны. Как правило, ПКП развивается в среднем или зрелом возрасте. Начало заболевания в раннем взрослом возрасте можно увидеть у людей с мутациями генов UROS или HFE. Начало в детстве встречается редко и м.б. связано с XT при онкологическом заболевании и с мутациями гена UROS.
P.S. * Окраска шифф-йодной кислотой.
2. Отклонения со стороны печени. ПКП почти всегда связана с неспецифическими нарушениями функции печени, особенно с повышением в сыворотке уровня трансаминаз и γ-глутамилтранспептидазы даже при отсутствии чрезмерного употребления алкоголя или гепатита С. Большинство гист. данных, таких как некроз, воспаление, гемосидероз и повышенное содержание жира, являются неспецифическими. К специфичным обнаружениям относятся красная флуоресценция ткани печени и флуоресцентные, игольчатые включения с двойным лучепреломлением, предположительно состоящие из порфиринов.
Электронная микроскопия показывает, что эти включения находятся в лизосомах, а паракристаллические включения обнаруживаются в митохондриях. При длительном течении заболевания чаще встречаются деформированная дольчатая архитектура и цирроз.
Риск развития гепатоцеллюлярной карциномы ↑, и частота встречаемости при ПКП составляет от 4 до 47%. Такие опухоли редко содержат большое количество порфиринов.
3. Другие признаки и взаимосвязи. Причины появления легкого или умеренного эритроцитоза у некоторых взрослых пациентов изучены недостаточно, но этим проявлениям способствуют хронические заболевания легких, вызванные курением. Более раннее появление симптомов м.б. отмечено у пациентов с генетическими предрасполагающими факторами, такими как унаследованный частичный дефицит УПДГ или генотип HFE C282Y/C282Y. Вторичный гемосидероз при таких заболеваниях, как миелофиброз и терминальная стадия почечной недостаточности, м.б. связан с ПКП.
Заболевание м.б. особенно тяжелым у пациентов с почечной недостаточностью, поскольку недостаток экскреции с мочой приводит к гораздо более высоким концентрациям порфиринов в плазме, а избыток порфиринов плохо поддается диализу. ПКП встречается чаще у пациентов с СКВ и др. иммунологическими нарушениями.
ж) Лабораторные признаки. При ПКП порфирины накапливаются в печени в основном в виде окисленных порфиринов, а не порфириногенов, на что указывает моментальная красная флуоресценция, наблюдаемая в ткани печени. Данный процесс развивается за несколько недель или месяцев, до того, как порфирины появляются в плазме и переносятся на кожу, вызывая фоточувствительность. В отличие от острой печеночной порфирии, для учета избытка порфиринов, выделяемых при ПКП, требуется лишь очень небольшое увеличение синтеза промежуточных продуктов гемового пути и незначительное или полное отсутствие увеличения печеночного ALAS1.
Дефицит УПДГ в печени приводит к сложной структуре избытка порфиринов, которые сначала накапливаются в виде порфириногенов, а затем подвергаются неферментативному окислению до соответствующих порфиринов (уро-, гепта-, гекса- и пентакарбоксилпорфиринов, а также изокопропорфиринов). В моче преобладают уропорфирин и гептакарбоксилпорфирин с меньшими количествами копропорфирина, пента- и гексакарбоксилпорфирина. Обычно вспомогательный путь выведения усиливается дефицитом УПДГ, при котором пентакарбоксилпорфириноген окисляется копропорфириногеноксидазой (СРОХ; следующий фермент в этом пути), образуя изокопропорфириноген — атипичный тетракарбоксилпорфириноген.
По отношению к нормальным показателям порфирины в моче повышены в большей степени, чем порфирины в кале. Но общее количество порфиринов, выделяемых с калом при ПКП, превышает таковое в моче, а общая экскреция изомеров типа III (включая изокопропорфирины, которые в основном происходят из серии типа III) превышает таковое из изомеров типа I. Возможно, поскольку уропорфириноген III является предпочтительным субстратом для УПДГ, уропорфириноген I накапливается больше, чем III, и выводится при ПКП в виде уропорфирина I. Гепта- и гексакарбоксил порфирин в основном являются изомером III, а пентакарбоксил порфирин и копропорфирин представляют собой смесь изомеров I и III в приблизительно равном соотношении.
з) Диагностика и дифференциальная диагностика. Определение общего порфирина в плазме является полезным для скрининга, т.к. при клинических признаках ПКП всегда ↑ плазменные порфирины. При его нормальных показателях исключаются ПКП и др. порфирии, вызывающие образование волдырей на коже. При ↑ уровня полезно определить максимум эмиссии флуоресценции плазмы при нейтральном pH, поскольку максимум ок. 619 нм характерен для ПКП (а также для ВЭП и НКП) и, что важно, исключает ВП, которая имеет явно отличающийся максимум флуоресценции.
Подтверждением является повышение уровня порфиринов в моче или плазме с преобладанием уропорфирина и гептакарбоксилпорфи-рина. Определение порфиринов мочи менее полезно для первоначального скрининга, поскольку неспецифическое повышение, особенно копропорфирина, происходит при заболеваниях печени и др. патологиях. Уровень АЛК в моче может незначительно ↑, а уровень ПБГ сохраняется в норме. Легкие случаи ВЭП могут клинически имитировать ПКП; так, эта возможность исключается при обнаружении нормальных или слегка повышенных уровней порфиринов эритроцитов.
Семейную ПКП (тип 2) можно отличить от спорадической (тип 1), обнаружив Ф активности УПДГ эритроцитов (при типе 2) или, что более надежно, мутацию УПДГ, связанную с заболеванием. Тип 3 отличается от типа 1 только наличием ПКП у родственника. Биохимические данные при ГЭП аналогичны таковым при ПКП, но с дополнительным заметным увеличением протопорфирина цинка в эритроцитах.
Псевдопорфирия (также известная как псевдо-ПКП) проявляется поражениями кожи, которые очень напоминают ПКП, но без значительного увеличения содержания порфиринов в плазме. Иногда задействован фотосенсибилизирующий агент, напр. НПВС. И ПКП, и псевдопорфирия могут возникать у пациентов с терминальной стадией почечной недостаточности.
и) Осложнения. Волдыри на коже могут лопнуть и инфицироваться, что иногда приводит к развитию флегмоны. При более тяжелом течении болезни у пациентов с терминальной стадией почечной недостаточности повторные инфекции могут приводить к увечьям, как при ВЭП. Псевдосклеродермия с рубцеванием, контрактурами и кальцификацией кожи и ПЖК является редким осложнением. Др. осложнения включают прогрессирующую болезнь печени и гепатоцеллюлярную карциному.
к) Лечение. Доступны две специфические и эффективные формы лечения: кровопускание и назначение низких доз гидроксихлорохина. По возможности следует исключить провоцирующие факторы. Для проведения лечения диагноз ПКП должен быть установлен очень точно, т.к. др. заболевания, вызывающие идентичные кожные поражения, не поддаются этим видам терапии. Лечение обычно можно начинать после обнаружения увеличения общего содержания порфиринов в плазме и исключения ВП с помощью анализа спектра флуоресценции при нейтральном pH, даже если еще нет данных по результатам исследования мочи и кала.
Следует прекратить употребление алкоголя, прием эстрогенов (женщинам) и курение; пациентам стоит пройти тестирование на мутации HCV, ВИЧ и HFE (белок человеческого регулятора гомеостатического железа). На выбор лечения влияют уровень и степень гемосидероза, оцениваемые по концентрации ферритина в сыворотке.
Кровопускание считается стандартной терапией и эффективно как у детей, так и у взрослых с ПКП, поскольку снижает содержание железа в печени. Лечение определяется уровнем ферритина и порфирина в плазме (или сыворотке). Для предотвращения симптоматической анемии необходимо следить за уровнем Hb или Ht. У взрослых единица крови (450 мл) удаляется с ~2-нед интервалами до тех пор, пока целевой уровень ферритина сыворотки не будет близок к нижнему пределу нормы (15 нг/мл).
Взрослым часто бывает достаточно проведения 6-8 кровопусканий. После этого уровень порфирина плазмы, составляющий до начала лечения 10-25 мкг/дл, продолжает постепенно ↓ до верхнего предела нормы (1 мкг/дл) в течение нескольких недель. Затем следует постепенное уменьшение кожных проявлений, иногда включая псевдосклеродермию. Может улучшиться состояние функции печени, исчезнут сидероз печени, игольчатые включения и красная флуоресценция ткани печени. Ремиссия обычно сохраняется, даже если уровень ферритина позже ↑, но рекомендуется следить за уровнем порфирина и повторно проводить кровопускания, если порфирины начинают ↑.
В случаях, когда кровопускания противопоказаны, могут использоваться инфузии дефероксамина — хелатора железа.
Альтернативой в случаях, когда кровопускания противопоказаны или плохо переносятся, является режим назначения низких доз гидроксихлорохина (или хлорохина). Нормальные дозы этих 4-аминохинолиновых противомалярийных ЛС при ПКП повышают уровни порфирина в плазме и моче и повышают фоточувствительность за счет усиления выведения порфиринов из печени. Это сопровождается острым гепатоцеллюлярным повреждением с лихорадкой, недомоганием, тошнотой и повышением уровня трансаминаз в сыворотке крови. Однако это сопровождается полной ремиссией порфирии.
Эти неблагоприятные последствия назначения нормальных доз в значительной степени можно избежать с помощью режима назначения низких доз (для взрослых, гидроксихлорохина 100 мг или хлорохина 125 мг, т.е. половины от обычной таблетки 2 р/нед), который можно продолжать до тех пор, пока уровни порфиринов в плазме или моче не нормализуются. Для маленьких детей рекомендуется назначение половины дозы взрослых. Прием гидроксихлорохина может ↓ риск развития ретинопатии. Механизм действия 4-аминохинолинов при ПКП неизвестен, но достаточно специфичен, поскольку эти ЛП не применяются при др. порфириях.
Недавние исследования показывают, что гидроксихлорохин в низких дозах так же безопасен и эффективен, как и проведение кровопускания у взрослых с ПКП.
У пациентов с ПКП и гепатитом С в первую очередь следует лечить ПКП, поскольку это состояние более симптоматично и лечится быстрее и эффективнее. Лечение ПКП с помощью кровопускания может оказаться невозможным, если лечение IFN-рибавирином осложняется анемией. Более того, лечение гепатита С может оказаться более эффективным после снижения уровня железа. Вопрос о том, следует ли первоначально использовать противовирусные ЛП прямого действия для лечения как гепатита С, так и ПКП, все еще изучается.
ПКП у пациентов с терминальной почечной недостаточностью часто протекает тяжелее и трудно поддается лечению. Однако введение эпоэтина бета («Эритропоэтина») может скорректировать анемию, мобилизовать железо и во многих случаях улучшить эффект от кровопускания. Улучшение после трансплантации почки м.б. частично связано с возобновлением эндогенной продукции эритропоэтина.
Для раннего выявления гепатоцеллюлярной карциномы всем пациентам с ПКП м.б. рекомендованы проведение инструментальных методов визуализация печени и определение сывороточного АФП, возможно, с интервалами продолжительностью 6-12 мес. Обнаружение низкой активности УПДГ эритроцитов или мутации UROS позволяет выявить заболевание тех, у кого есть основная генетическая предрасположенность; это не оказывает влияния на лечение, но полезно для генетического консультирования.
л) Прогноз. Поздняя кожная порфирия является наиболее легко поддающейся лечению формой порфирий. Полная ремиссия возможна при лечении либо проведением кровопусканий, либо назначением низких доз гидроксихлорохина. В настоящий момент имеется мало информации о частоте рецидивов и долгосрочной перспективе. Риск развития гепатоцеллюлярной карциномы ↑, а некоторые провоцирующие факторы, такие как гепатит С, могут привести к осложнениям даже после того, как ПКП будет находиться в стадии ремиссии.
м) Профилактика и генетическое консультирование. Наследственную мутацию UROS обычно можно обнаружить или исключить путем измерения активности УПДГ эритроцитов, хотя исследования ДНК более чувствительны. Родственники пациентов с мутациями UROS имеют повышенный риск развития ПКП и могут иметь повышенную мотивацию избегать нежелательных моделей поведения, таких как употребление этанола и табака, а также воздействия HCV и ВИЧ (хотя такое консультирование м.б. предоставлено любому). Обнаружение мутаций HFE, особенно C282Y, должно вызвать необходимость проведения скрининга среди родственников (некоторые из них м.б. гомозиготами по C282Y) и потребовать пожизненного мониторинга сывороточного ферритина.