Пиримидины являются структурными блоками ДНК и РНК и участвуют в образовании активных промежуточных продуктов в метаболизме углеводов и фосфолипидов (напр., уридиндифосфат глюкозы, цитидиндифосфат холина), глюкуронизации в процессах детоксикации (уридиндифосфат) и гликозилировании белков и липидов.
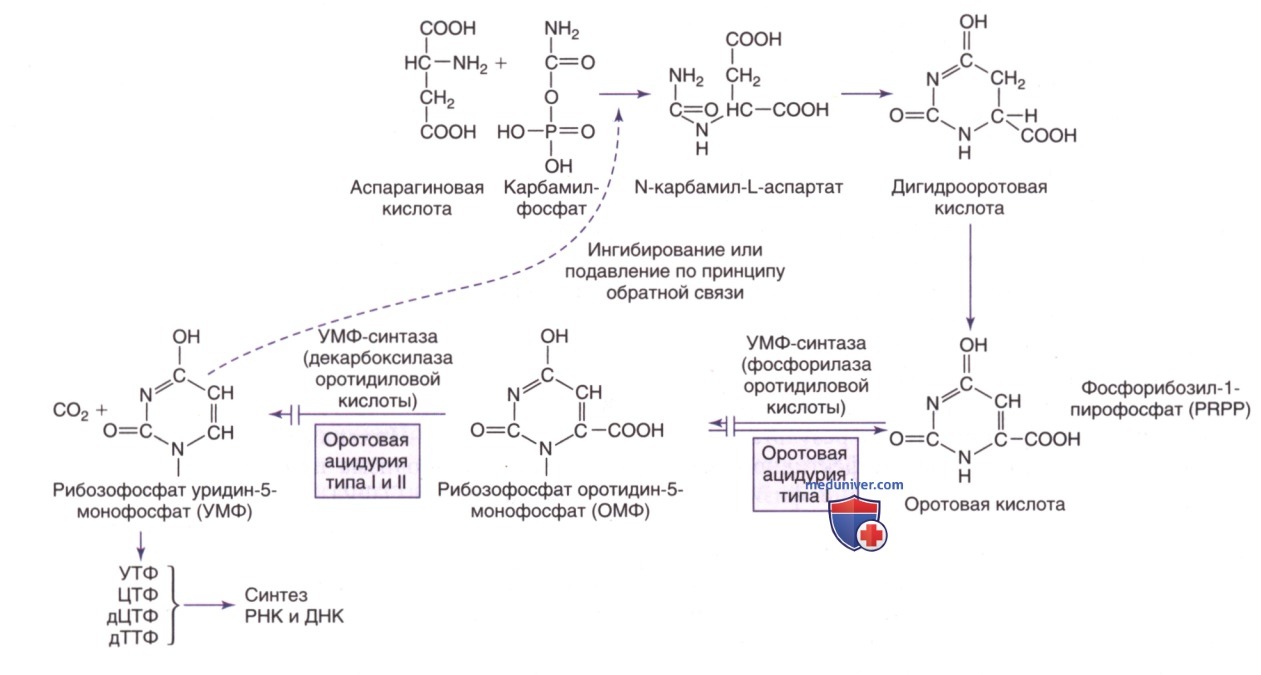
Основным предшественником биосинтеза пиримидина является карбамилфосфат, участвующий в цикле мочевины. Следовательно, проксимальные блокировки цикла мочевины приводят к избыточному направлению карбамилфосфата в цикле обмена пиримидина. Синтез пиримидина отличается от синтеза пуринов тем, что сначала образуется оротовая кислота с одинарным пиримидиновым кольцом, которая затем связывается с рибозофосфатом с образованием центрального пиримидиннуклеотида уридинмонофосфата (УМФ).
Пиримидиновые основания (урацил и тимин) катаболизируются в четыре этапа (см. рис. ниже). Ниже описаны восемь нарушений обмена пиримидина. Катаболизм пуринов имеет конечную точку, легко измеряемую по мочевой кислоте; однако в катаболизме пиримидина нет эквивалентного соединения. Нарушения синтеза пиримидина de novo включают наследственную оротовую ацидурию и дефицит дигидрооротатдегидрогеназы (синдром Миллера). Дефицит тимидинкиназы является частью утилизации пиримидина, а др. нарушения связаны с гиперактивностью (при одном синдроме) или дефектами пути разложения пиримидина.
Пути биосинтеза пиримидина. PRPP — фосфорибозилпирофосфат
Нарушения обмена пиримидина могут проявляться в виде анемии, невропатологии, акрофациального дизостоза или мультисистемных митохондриальных нарушений. Первые три этапа путей разложения тимина и урацила, соответственно, используют одни и те же ферменты [DPD, DPH и уреидопропионаза (UP)]. В результате указанных трех этапов урацил превращается в β-аланин. Появляется все больше подтверждений тому, что пиримидины играют важную роль в регуляции НС. Предполагается, что при снижении нейромедиаторной функции β-аланина появляются клинические симптомы. Клинически нарушения обмена пиримидина могут остаться незамеченными, поскольку они являются редкими, а их симптомы не очень специфичны.
Тем не менее их следует рассматривать как возможные причины анемии и неврологических заболеваний. Также они являются противопоказанием для лечения онкологических больных некоторыми аналогами пиримидина.
а) Дефицит уридин-монофосфат-синтазы 1-го типа (наследственная оротовая ацидурия). Наследственная оротовая ацидурия представляет собой нарушение синтеза пиримидина, связанное с недостаточной активностью двух последних стадий пути синтеза пиримидина de novo: оротатфосфорибозилтрансферазы и оротидин-5’-монофосфатдекарбоксилазы. Активность двух указанных этапов находится в отдельных доменах бифункционального белка, УМФ-синтазы. Этот белок катализирует двухстадийное превращение оротовой кислоты в УМФ через монофосфат оротидина. Наследственная оротовая ацидурия (дефицит УФМ-синтазы) приводит к чрезмерному накоплению оротовой кислоты.
1. Генетика. Дефицит УМФ-синтазы наследуется по АуР-типу, при этом оба функциональных домена кодируются одним геном УМФС, расположенным на длинном плече хромосомы 3 (3q13). Теоретически случайные мутации в гене имеют равную вероятность образования оротатфосфорибозилтрансферазы или дефицита оротидин-5’-монофосфатдекарбоксилазы, однако был зарегистрирован только один случай дефицита оротидин-5’-монофосфатдекарбоксилазы. Генетические метаболические дефекты, в которых участвуют четыре из шести ферментов, связанных с циклом мочевины, также могут приводить к оротовой ацидурии, вторичной по отношению к истощению PPRP, возникающему в результате значительного увеличения синтеза пиримидина.
2. Клинические проявления. У пациентов с наследственной оротовой ацидурией (дефицит УМФ-синтазы 1-го типа) наблюдается макроцитарная гипохромная мегалобластная анемия, не поддающаяся обычной терапии (железо, фолиевая кислота, витамин В12), у таких пациентов возможно развитие лейкопении. Заболевание обычно проявляется в первый месяц жизни. При отсутствии лечения это нарушение может привести к умственной отсталости, задержке физического развития, сердечным заболеваниям, косоглазию, кристаллурии и периодической обструкции мочеточника.
Функция почек обычно в норме. У гетерозиготных пациентов может наблюдаться оротовая ацидурия в легкой форме, но др. поражений нет. Считается, что клинические признаки связаны с истощением пиримидиновых нуклеотидов. Метаболиты, полученные из нескольких фармакологических агентов (напр., 5-азауридина, аллопуринола), могут вызывать вторичную оротовую ацидурию и оротидинурию, специфически ингибируя стадию оротидин-5’-монофосфатдекарбоксилазы УМФ-синтазы. Развитие оротовой ацидурии также м.б. связано с парентеральным питанием, дефицитом незаменимых аминокислот и синдромом Рея.
3. Лабораторные признаки. Ферментативный дефект может проявляться в печени, лимфобластах, эритроцитах, лейкоцитах и культуре фибробластов кожи. Доступен тест на выявление носителя, а также пренатальная диагностика.
4. Лечение. Введение уридина в дозах 50-300 мг/кг в сутки привело к клиническому улучшению и снижению экскреции оротовой кислоты при дефиците УМФ-синтазы 1-го типа. Лечение необходимо на протяжении всей жизни. Урацил оказывается неэффективным, поскольку, в отличие от пуринов, восстановление пиримидина происходит на уровне нуклеозидов (уридина). Долгосрочный прогноз в неосложненных случаях является благоприятным; однако врожденные пороки развития и др. связанные с ними особенности могут отрицательно повлиять на исход заболевания.
б) Дефицит дигидро-оротат-дегидрогеназы (синдром Миллера). Синдром Миллера был первым менделевским нарушением, молекулярная основа которого идентифицирована путем секвенирования всего экзома. Было показано, что это нарушение коррелирует с мутациями в дигидрооро-татдегидрогеназе. Фермент дигидрооротатдегидрогеназа связан с митохондриальной цепью переноса электронов и катализирует окисление дигидрооротата до оротовой кислоты, что необходимо для синтеза пиримидина de novo.
1. Клинические проявления. Синдром Миллера — это узнаваемый синдром акро-фациального дизостоза с сочетанием черепно-лицевых аномалий и аномалий конечностей. Он включает микрогнатию, ротолицевые расщелины, гипоплазию скуловой кости, аплазию центральных ресниц нижнего века, расщелину губы или нёба, колобому нижнего века и чашевидные уши в сочетании с постаксиальной деформацией конечностей, гипоплазию конечностей с локтевым суставом или без него, гипоплазию малоберцовой кости и лишние соски. Многие из этих признаков похожи на синдром Тричера-Коллинза.
2. Лабораторные признаки. Анализы ассоциированных с заболеванием аллелей дигидрооротатдегидрогеназы показывают, что люди с этой патологией имеют дефицит синтеза пиримидина de novo, но при этом сохраняется значительная остаточная функция.
3. Лечение. Согласно теоретическим представлениям, пищевые добавки оротовой кислоты или уридина должны обходить метаболический блок. Однако поскольку основные эффекты происходят в утробе матери, маловероятно, что фенотипические отклонения можно исправить.
в) Дефицит дигидро-пиримидин-дегидрогеназы (тиминурацилурия, пиримидинурия). DPD катализирует начальную и лимитирующую стадию разложения пиримидиновых оснований урацила и тимина. DPD выявлен в большинстве тканей, с наибольшей активностью в лимфоцитах.
1. Генетика. Дефицит DPD представляет собой АуР-заболевание, при котором ген DPYD расположен в хромосоме 1р22, при этом обнаружено не менее 32 полиморфизмов. По оценкам, частота гетерозиготности может достигать 3%.
2. Клинические проявления. У детей могут развиваться судороги, умственная отсталость и задержка развития моторики. Менее распространенными признаками являются задержка роста, микроцефалия, аутистическое поведение и глазные аномалии. У др. могут быть более легкие неврологические симптомы и нарушение речи. Сообщалось о пациентах с этой патологией без поражений, что предположительно указывает на возможные вторичные генные эффекты. Большинство пациентов имеют начальный период нормального психомоторного развития, за которым следует задержка в развитии. Симптомы м.б. связаны с изменением гомеостаза урацила, тимина или β-аланина.
Поскольку β-аланин является структурным аналогом γ-аминомасляной кислоты и глицина, предполагалось, что он может влиять на ингибирующую нейропередачу. DPD является первичным и лимитирующим ферментом в инактивации противоопухолевого препарата 5-фторурацила (5-ФУ), ответственного за 80% его катаболизма. Пациенты с частичным дефицитом DPD подвержены риску развития тяжелой токсичности, связанной с 5-ФУ. У взрослых, ранее здоровых пациентов после приема 5-ФУ для лечения рака наблюдалась нейротоксичность (головная боль, сонливость, зрительные иллюзии, нарушение памяти), связанная с пиримидинемией.
3. Лабораторные признаки. Дефицит DPD характеризуется переменным фенотипом и диагностируется по обширному накоплению тимина и урацила в моче (тимин-урацилурия), плазме и СМЖ. Уровень мочевой кислоты соответствует норме. Зарегистрированы случаи пренатального установления диагноза.
4. Лечение. Отсутствует утвержденное лечение этого нарушения, хотя пациенты с судорогами поддаются лечению противосудорожными ЛП. Генетические варианты DPYD, связанные с частичной или полной активностью DPD и встречающиеся относительно часто в популяциях, являются потенциально полезными прогностическими маркерами ответа пациента на XT 5-ФУ
г) Дефицит дигидропиримидиназы (дигидропиримидинурия). Дефицит дигидропиримидиназы (DPH; англ. dihydropyrimidinase) является вторым ферментом в трехступенчатом пути разложения урацила и тимина. Дефицит DPH характеризуется повышенной экскрецией с мочой дигидроурацила и дигидротимина (дигидропиримидинурия), а также урацила и тимина. Подобно дефициту DPD, существуют вариации клинического фенотипа.
1. Генетика. Это АуР-заболевание, при котором ген DPYS расположен в хромосоме 8q22. Исследования не обнаружили значительных различий в остаточной активности между мутациями, наблюдаемыми у симптомных и бессимптомных пациентов, что похоже на дефицит DPD. Частота встречаемости среди населения Японии составила 0,1%.
2. Клинические проявления. Клинические проявления аналогичны дефициту DPD, и это подтверждает, что дефекты этих последовательных этапов вызывают общее заболевание. Симптомы в трех несвязанных случаях включали судороги с дисморфическими признаками и задержку развития у двух пациентов. Однако в рамках проводившейся в Японии скрининговой программы, направленной на выявление нарушений деградации пиримидинов, было выявлено три случая заболевания у младенцев, не являвшихся родственниками, а также два случая у взрослых. Все эти случаи были бессимптомными, несмотря на накопление продуктов деградации пиримидинов в жидкостях организма.
3. Лабораторные признаки. Скрининг на содержание органических кислот позволяет выявить повышенное количество урацила и тимина в моче. Для выявления носителей дефицита DPH использовались пероральные нагрузочные тесты с урацилом, дигидроурацилом, тимином и дигидротимином. В симптоматических случаях попытки лечения β-аланином дали сомнительные результаты. Зарегистрирован один случай повышенной чувствительности к 5-ФУ.
д) Дефицит β-уреидопропионазы (N-карбамил-β-аминоацидурия). В результате последовательного действия трех ферментов пиримидиновые основания урацил и тимин подвергаются разложению до β-аланина и β-аминоизомасляной кислоты соответственно. Третьим ферментом в этом пути является уреидопропионаза, и ее дефицит приводит к N-карбамил-β-аминоацидурии. 3-уреидопропионовая кислота действует как эндогенный нейротоксин за счет ингибирования митохондриального энергетического метаболизма, что приводит к запуску вторичных энергозависимых эксайтотоксических механизмов.
1. Генетика. Флуоресцентная гибридизация in situ (FISH) локализует ген β-уреидопропионазы человека UPB1 в хромосоме 22q11.2.
2. Клинические проявления. К клиническим проявлениям относятся мышечная гипотония, дистонические движения, судороги и серьезная задержка развития. Сообщалось о некоторых людях с дефицитом уреидопропионазы и неврологическими проблемами.
3. Лабораторные признаки. Невропатология затрагивает как серое, так и белое в-во. Дефицит УП приводит к патологическому накоплению 3-3-уреидопропионовая кислоты в физиол. жидкостях. Анализ мочи в зарегистрированном случае показал повышенные уровни N-карбамил-β-аланина и N-карбамил-β-аминоизомасляной кислоты (уреидоизомасляной кислоты). Фермент экспрессируется только в печени, и при биопсии печени активность β-уреидопропионазы не обнаруживается.
е) Дефицит пиримидин-5'-нуклеотидазы. Созревание эритроцитов сопровождается распадом РНК и высвобождением мононуклеотидов. Пиримидин-5’-нуклеотидаза является первым ферментом разложения пиримидинового цикла восстановления и катализирует гидролиз пиримидиновых 5’-нуклеотидов до соответствующих нуклеозидов. Дефицит фермента приводит к накоплению высоких концентраций цитидиновых и уридиновых нуклеотидов в эритроцитах, что, в свою очередь, приводит к гемолизу. Дефицит пиримидин-5’-нуклеотидазы может, по крайней мере, частично компенсироваться in vivo др. нуклеотидазами или, возможно, др. путями метаболизма нуклеотидов.
1. Генетика. Это АуР-заболевание с участием гена NT5C3A, расположенного в хромосоме 7 (7p15).
2. Клинические проявления. Пациенты с дефицитом пиримидин-5’-нуклеотидазы клинически имеют дефект, ограниченный эритроцитами и характеризующийся несфероцитарной гемолитической анемией с базофильной зернистостью. Др. характерными признаками являются спленомегалия, повышенный уровень непрямого билирубина и гемоглобинурия. Свинец является мощным ингибитором пиримидин-5’-нуклеотидазы, поэтому оценка содержания свинца должна проводиться каждый раз, когда одновременно обнаруживаются гемолитическая анемия, дефицит пиримидин-5’-нуклеотидазы и базофильная зернистость.
3. Лабораторные признаки. Для диагностики необходимо проведение анализа гидролиза УМФ в эритроцитах с образованием уридина и неорганического фосфата. Пациентов с несфероцитарной гемолитической анемией с базофильной зернистостью следует обследовать на ферментный дефект. Анемия обычно умеренная, и переливание крови требуется редко.
4. Лечение. Специфическое лечение отсутствует. Спленэктомия оказалась неэффективной. Дефицит пиримидин-5’-нуклеотидазы, приобретенный вследствие воздействия свинца, поддается лечению, в отличие от врожденного дефицита.
ж) Повышенная активность цитозольной 5'-нуклеотидазы (истощение пиримидиновых нуклеотидов). Истощение пиримидиновых нуклеотидов и повышенная активность цитозольной 5’-нуклеотидазы могут привести к нарушению нервно-психического развития. У четырех пациентов, не являющихся родственниками, было обнаружено 6-10-кратное повышение активности пиримидин-5’-нуклеотидазы в фибробластах как с пуриновыми, так и с пиримидиновыми субстратами. Исследование культуры фибробластов этих пациентов показало нормальное включение пуриновых оснований в нуклеотиды, но пониженное включение уридина и оротовой кислоты.
1. Клинические проявления. В течение первых нескольких лет жизни появляются такие клинические симптомы, как задержка психического развития, судороги, атаксия, рецидивирующие инфекции, серьезная задержка речевого развития, гиперактивность, низкая концентрация внимания и агрессивное поведение. У пациентов с этой патологией на ЭЭГ наблюдаются аномалии. При метаболическом исследовании показатели соответствуют норме, за исключением стойкой гипоурикозурии. Предполагается, что дефицит пиримидиновых нуклеотидов возникает по причине повышенной катаболической активности и пониженной утилизации пиримидина.
2. Лечение. Пероральный прием уридина направлен на компенсацию повышенного катаболизма нуклеотидов. У всех зарегистрированных пациентов, получавших уридин, наблюдалось улучшение речи и поведения, снижение судорожной активности после прекращения приема противосудорожных ЛП и снижение восприимчивости к инфекциям.
з) Дефицит тимидин-фосфорилазы (митохондриальная нейрогастроинтестинальная энцефаломиопатия). Тимидинфосфорилаза катализирует катаболизм тимидина до тимина. Этот фермент также известен как тромбоцитарный фактор роста эндотелиальных клеток благодаря его ангиогенным св-вам или глиостатин, что подтверждает его ингибирующее действие на пролиферацию глиальных клеток. Он участвует в метаболизме митохондриальных нуклеозидов. Содержание тимидина в плазме пациентов с этой патологией повышено более чем в 20 раз по сравнению с контрольной группой.
Потеря функции тимидинфосфорилазы приводит к митохондриальной нейрогастроинтестинальной энцефаломиопатии (МНГИЭ), которая наследуется как единичное АуР-заболевание, вызывая истощение и нестабильность митохондриальной ДНК. При МНГИЭ потеря активности тимидинфосфорилазы приводит к токсическим накоплениям нуклеозидов тимидина и дезоксиуридина, которые фосфорилируются до соответствующих нуклеозидтрифосфатов в митохондриях. Это приводит к дисбалансу пула дезоксинуклеозидтрифосфата митохондрий и аберрантной репликации митохондриальной ДНК.
1. Генетика. Ген TYMP, кодирующий тимидинфосфорилазу, идентифицирован как ген МНГИЭ и расположен в хромосоме 22q13.32-qter, но белок внедряется в митохондрии.
2. Клинические проявления. Клинические проявления МНГИЭ обычно начинаются в подростковом и юношеском возрасте и включают птоз, прогрессирующий внешний офтальмопарез, нарушение моторики ЖКТ (псевдообструкцию) и мальабсорбцию, кахексию, периферическую невропатию, миопатию скелетных мышц и лейкоэнцефалопатию.
3. Лабораторные признаки. При проведении биопсии мышц обычно выявляются митохондриальные аномалии. Для скрининга используют метод обнаружения тимидина и дезоксиуридина в моче и плазме, которые в норме отсутствуют, но значительно повышены при этой патологии. Диагноз подтверждают путем анализа активности тимидинфосфорилазы в периферических лейкоцитах. Молекулярно-генетический анализ выявляет функциональные мутации в гене TYMP. Повышенное содержание нуклеотидов тимидина и/или дезоксиуридина может вызвать дисбаланс пула митохондриальных нуклеотидов. Это приводит к изменениям митохондриальной ДНК, в частности к истощению ДНК.
4. Лечение. Показана поддерживающая терапия. Лечение МНГИЭ не разработано. Нескольким пациентам была выполнена трансплантация костного мозга, но об улучшении симптомов или прогрессировании заболевания не сообщалось. Потенциальным лечением МНГИЭ является трансплантация аллогенных стволовых кроветворных клеток для восстановления активности тимидинфосфорилазы и удаления токсичных метаболитов.
и) Дефицит тимидинкиназы-2. Тимидинкиназа-2 является ключевым ферментом в пути восстановления пиримидина, обеспечивающим образование предшественника нуклеотида для митохондриальной ДНК. Дефицит тимидинкиназы-2 приводит к тканеспецифическому истощению митохондриальной ДНК. В норме тимидинкиназа-2 фосфорилирует тимидин и дезоксицитидин.
1. Генетика. Ген тимидинкиназы-2 расположен в хромосоме 16q 22; дефицит наследуется по АуР-типу.
2. Клинические проявления. У людей с дефицитом тимидинкиназы-2 в младенчестве наблюдается тяжелая миопатия и истощение мышечной митохондриальной ДНК.
3. Лечение. Специального лечения не разработано. Показана поддерживающая терапия.