Молоко и молочные продукты содержат лактозу — главный диетический источник галактозы. В результате метаболизма галактозы производится энергия для клеточного метаболизма за счет ее преобразования в глюкозо-1-фосфат (см. табл. 1). Галактоза также играет важную роль в образовании галактозидов, включая гликопротеины, гликолипиды и гликозаминогликаны.
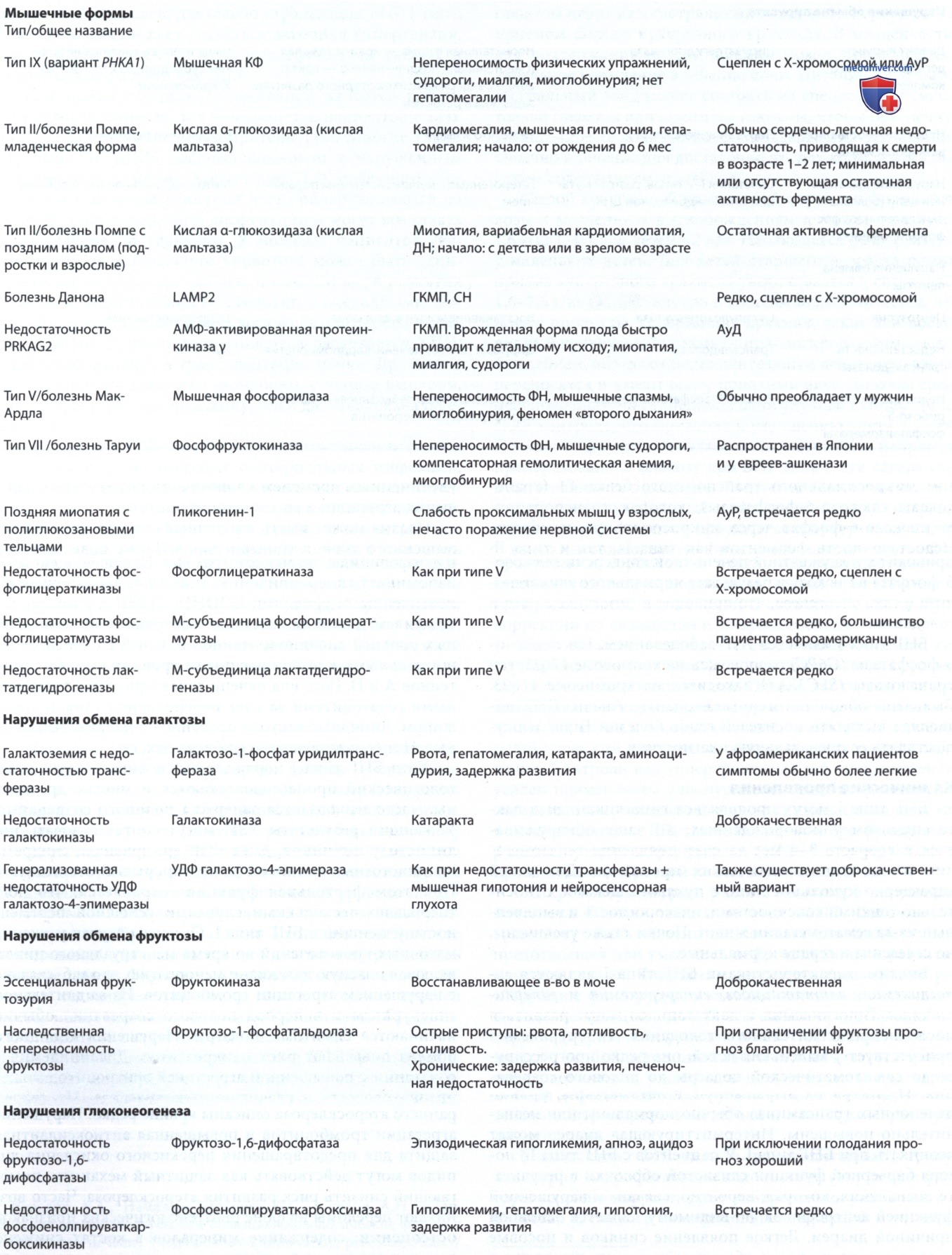
Галактоземия означает повышенный уровень галактозы в крови и обнаруживается в трех разл. врожденных дефектах метаболизма галактозы в одном из следующих ферментов: галактозо-1-фосфатуридилтрансфераза, галактокиназа и УДФ галактоза-4-эпимераза. Термин «галактоземия», хотя и соответствует дефициту при любом из этих расстройств, обычно обозначает дефицит трансферазы.
а) Недостаточность галактозо-1-фосфатуридилтрансферазы при галактоземии. Существует две формы недостаточности: младенцы с полной или почти полной недостаточностью фермента (классическая галактоземия) и дети с частичной недостаточностью трансферазы.
Классическая галактоземия — серьезное заболевание, симптомы которого обычно проявляются во 2-й половине 1-й недели жизни. Прогнозируемая частота встречаемости составляет 1:60 000 живорожденных. Новорожденный ребенок получает большое количество лактозы (до 40% в грудном молоке и некоторых смесях), которая состоит из равных частей глюкозы и галактозы. Без фермента трансферазы младенец не может метаболизировать галактозо-1-фосфат, накопление которого приводит к повреждению почек, печени и мозга.
В результате трансплацентарного поступления галактозы из пищи, употребляемой гетерозиготной матерью, или эндогенной продукции галактозы самим эмбрионом вышеперечисленные нарушения возникают у пораженного плода в/утробно.
РФ: в основу современной классификации галактоземии положен этиологический принцип. Существует три варианта галактоземии, в зависимости от имеющегося у больного дефекта одного из трех основных ферментов, принимающих участие в метаболизме глюкозы.
• Галактоземия I типа — дефицит галактозо-1-фосфатуридилтрансферазы, обычно сопровождается тотальной мальабсорбцией галактозы и приводит к возникновению наиболее тяжелых форм заболевания.
• Галактоземия II типа — дефицит фермента галактокиназы.
• Галактоземия III типа — дефицит фермента уридин-дифосфат-галактозо-4-эпимеразы*.
P.S. * КР «Нарушения обмена галактозы (галактоземия)», 2021 г.
1. Клинические проявления. Диагноз дефицита уридилтрансферазы необходимо исключать у новорожденных или детей грудного возраста первых месяцев жизни при проявлении в течение нескольких дней или недель после рождения любых из следующих признаков: желтуха, гепатомегалия, рвота, гипогликемия, судороги, летаргия, раздражительность, трудности с кормлением, недостаточное увеличение МТ или неспособность восстановить МТР и аминоацидурия. У детей, не получающих лечения, могут наблюдаться ядерная катаракта, кровоизлияние в стекловидное тело, печеночная недостаточность, цирроз, асцит, спленомегалия или умственная отсталость.
Пациенты с галактоземией подвергаются повышенному риску неонатального сепсиса, вызванного кишечной палочкой; начало сепсиса часто предшествует диагнозу галактоземии. Может возникнуть псевдоопухоль головного мозга (доброкачественная внутричерепная гипертензия)*, вызывающая выбухание родничка. Полное исключение лактозы из рациона приводит к уменьшению острых симптомов. При отсутствии лечения смерть от печеночной, почечной недостаточности и сепсиса может наступить в течение нескольких дней. Если диагноз не поставлен при рождении, поражения печени (цирроз печени) и ГМ (умственная отсталость) становится все более тяжелыми и необратимыми.
P.S. * Доброкачественной внутричерепной гипертензией (ДВГ) или псевдоопухолью мозга называется первичная внутричерепная гипертензия, обусловленная нарушением динамики СМЖ. При этом отсутствует внутричерепное объемное образование и гидроцефалия.
Частичная недостаточность трансферазы обычно протекает бессимптомно. Она более распространена, чем классическая галактоземия, и диагностируется при скрининге новорожденных из-за умеренно повышенной галактозы в крови и/или низкой активности трансферазы. Галактоземию следует заподозрить у новорожденных или детей грудного возраста, которые отстают в развитии или у которых есть какие-либо из предыдущих признаков. Световая и электронная микроскопия ткани печени выявляет жировую инфильтрацию, формирование псевдоацинусов и возможный макронодулярный цирроз. Такие изменения соответствуют нарушениям обмена в-в, но не указывают на точную ферментативную недостаточность.
2. Диагностика. Первичный диагноз галактоземии ставится при обнаружении восстанавливающего в-ва в нескольких образцах мочи, собранных, когда пациент получает питание, содержащее женское молоко, коровье молоко или любую др. смесь, включающую лактозу. Восстанавливающее в-во, обнаруживаемое Clinitest в моче (напр., глюкоза, галактоза), м.б. идентифицировано с помощью хроматографии или ферментативного теста, специфичного для галактозы. Галактоза м.б. обнаружена в моче при условии, что кормление молоком проводилось в течение последних нескольких часов и у ребенка нет выраженной рвоты.
Результаты анализа мочи Clinistix обычно отрицательны, поскольку тест основан на действии глюкозооксидазы, которая специфична для глюкозы, но не реагирует с галактозой. Аминокислоты м.б. обнаружены в моче, так как они выводятся вместе с глюкозой из-за наличия синдрома проксимальных почечных канальцев.
Поскольку галактоза опасна для людей с галактоземией, не следует использовать диагностические контрольные тесты, зависящие от введения галактозы внутрь или в/в. Диагноз можно установить методом прямого ферментного анализа с использованием эритроцитов.
Диагностика основывается на определении низкого уровня активности фермента галактозо-1-фосфатуридилтрансферазы в эритроцитах крови. Поскольку активность фермента определяют в эритроцитах, нецелесообразно выполнение исследования в течение как минимум одного месяца после переливания плазмы крови человека (эритроцитарной массы).
Клиницист должен подтвердить, что пациенту не делали переливание крови до взятия образца крови, поскольку можно упустить правильный диагноз. Современный метод использует нерадиоактивное УФ-излучение и высокоэффективную жидкостную хроматографию для точного определения уровней галактозо-1-фосфатуридилтрансферазы в эритроцитах.
3. Генетика. Дефицит трансферазы является АуР-заболеванием. По данным скрининга новорожденных в США, частота заболевания составляет 1:47 000 живорождений. Существует несколько ферментативных вариантов галактоземии. Вариант Дуарте с заменой одной аминокислоты (p.N314D) отличается пониженной активностью фермента эритроцитов (50% от нормы), но обычно не имеет клинического значения. Этот вариант является наиболее распространенным с несущей частотой 12% среди населения в целом. Те, кто является гетерозиготным по варианту Дуарте галактоземии, обычно имеют 25% нормальной активности галактозы, невыраженную симптоматику, повышенный уровень метаболитов и не нуждаются в лечении.
Др. подобные варианты, проявляющие небольшую активность фермента, обычно не требуют вмешательства. Некоторые пациенты афроамер. происхождения имеют более легкие симптомы, несмотря на отсутствие измеримой активности трансферазы в эритроцитах; у этих пациентов сохраняется 10% активность ферментов в печени и слизистой оболочке кишечника, в то время как у большинства пациентов европеоидной расы активность ни в одной из этих тканей не обнаруживается. Более 230 идентифицируемых патогенных вариантов связаны с дефицитом трансферазы. У афроамериканцев 62% аллелей представлены вариантом p.S135L, который отвечает за более легкое течение болезни.
В белом населении 70% аллелей представлены миссенс-вариантами p.Q188R и p.K285N и связаны с тяжелым заболеванием. Тестирование носительства и пренатальная диагностика м.б. выполнены путем прямого ферментативного анализа амниоцитов или ворсинок хориона; тестирование также м.б. основано на ДНК.
4. Лечение и прогноз. С появлением неонатального скрининга на галактоземию стало возможным выявлять и лечить на более ранних этапах. Все продукты, содержащие галактозу, следует исключить из рациона при первоначальном подозрении на галактоземию. Доступны разл. заменители молока, не содержащие лактозу (гидролизаты казеина, смеси на основе сои). Исключение галактозы из рациона вместе с адекватным добавлением кальция устраняет задержку роста, а также почечную и печеночную дисфункцию.
Катаракта регрессирует, и у большинства пациентов не наблюдается ухудшения зрения. Ранняя диагностика и лечение продемонстрировали улучшение прогноза галактоземии. Однако при длительном наблюдении пациенты по-прежнему проявляют недостаточность яичников с первичной или вторичной аменореей, снижением минеральной плотности костей, задержкой развития и трудностями в обучении, которые усиливаются с возрастом. Гипергонадотропный гипогонадизм наблюдается у 80-90% пациенток с классической галактоземией. Хотя большинство женщин с классической галактоземией становятся бесплодными по достижении детородного возраста, небольшое число женщин все же родили.
У большинства пациентов наблюдаются нарушения речи, у меньшего — замедленный рост, нарушение двигательной функции и равновесия (с явной атаксией или без нее). Относительный контроль уровней галактозо-1-фосфата не всегда коррелирует с долгосрочным исходом, что приводит к мысли, что за это м.б. ответственны др. факторы, такие как повышенный уровень галактитола, пониженный уровень УДФ-галактозы (донор для галактолипидов и белков) и выработка эндогенной галактозы.
б) Недостаточность галактокиназы. Недостаточный фермент — галактокиназа, которая обычно катализирует фосфорилирование галактозы. Основными накопленными метаболитами являются галактоза и галактитол. Сообщается, что два гена кодируют галактокиназу: GK1 на хромосоме 17q24 и GK2 на хромосоме 15. Катаракта обычно считается единственным проявлением недостаточности галактокиназы; псевдоопухоль мозжечка является редким осложнением. В остальном болезнь у младенца протекает бессимптомно. У гетерозиготных носителей м.б. риск возрастной катаракты.
Результаты лабораторных исследований показывают повышенную концентрацию галактозы в крови при условии, что младенец получал смесь, содержащую лактозу. Диагноз ставится на основании подтверждения отсутствия активности галактокиназы в эритроцитах или фибробластах. Активность трансферазы в норме. Лечение — ограничение галактозы в диете
в) Недостаточность уридин-дифосфат-галактозо-4-эпимеразы. Есть два разл. формы недостаточности эпимеразы. Первая — доброкачественная форма, которая диагностируется случайно в рамках программ скрининга новорожденных. У заболевших пациентов течение бессимптомное, поскольку недостаточность ферментов ограничивается лейкоцитами и эритроцитами. Эта форма не требует лечения. Вторая разновидность является тяжелой, потому что больше распространена недостаточность эпимеразы.
Клинические проявления напоминают недостаточность трансферазы с дополнительными симптомами гипотонии и нейрогенной тугоухости. Клинические симптомы улучшаются при ограничении галактозы в диете. Хотя тяжелая форма галактоземии встречается редко, ее следует рассматривать у пациента с симптомами, имеющего поддающийся измерению галактозо-1-фосфат и нормальную активность трансферазы. Аномально накопленные метаболиты аналогичны метаболитам при недостаточности трансферазы; тем не менее также наблюдается увеличение клеточный УДФ-галактозы. Диагноз подтверждается определением эпимеразы в эритроцитах.
Пациенты с тяжелой формой недостаточности эпимеразы не могут синтезировать УДФ-галактозу из УДФ-глюкозы и зависят от галактозы. Поскольку галактоза является важным компонентом многих структурных белков нервной системы, пациентам определяют диету с ограничением галактозы, а не диету без галактозы.
Младенцы с легкой формой недостаточности эпимеразы не нуждаются в лечении. Желательно следить за образцами мочи на предмет редуцирующих в-в и исключить аминоацидурию в течение нескольких недель после постановки диагноза, пока ребенок все еще находится на лактозосодержащей смеси.
Ген УДФ-галактозо-4-эпимеразы (GALE) расположен на хромосоме 1 на 1р36. Обнаружение носителя возможно путем измерения активности эпимеразы в эритроцитах. Пренатальная диагностика тяжелой формы недостаточности эпимеразы м.б. выполнена с помощью ферментативного анализа культивированных клеток околоплодных вод.