Нарушения расщепления и структуры гликопротеинов включают несколько лизосомных болезней накопления, которые возникают в результате дефектов расщепления гликопротеинов, а также врожденных нарушений гликозилирования. Гликопротеины — это макромолекулы, в которых к белковому остову присоединены олигосахаридные цепи. Синтез их происходит двумя путями: гликозилтрансферазным путем, в ходе которого олигосахариды соединяются О-гликозидными связями с остатками серина или треонина в белке; и долихололовым, липид-связанным путем, при котором олигосахаридные цепи соединяются N-гликозидными связями с амидной группой аспарагина.
Лизосомные болезни накопления гликопротеинов возникают в результате дефицита ферментов, которые обычно участвуют в разрушении олигосахаридов и включают сиалидоз, галактосиалидоз, аспартилглюкозаминурию и α-маннозидоз. В некоторых случаях лежащая в основе аномалия, которая приводит к накоплению гликопротеинов, также приводит к аномальному распаду др. классов макромолекул, которые содержат аналогичные олигосахаридные связи (некоторые гликолипиды и протеогликаны). В этих случаях основная ферментативная недостаточность приводит к накоплению как гликопротеинов, так и гликолипидов.
Классификация этих типов нарушений как липидозов или гликопротеинозов зависит от природы преимущественно накапливаемых в-в. В целом гликопротеиновые расстройства характеризуются АуР-наследованием и прогрессирующим течением заболевания с клиническими признаками, сходными с мукополисахаридозами.
а) Сиалидоз и галактосиалидоз. Сиалидоз — это АуР-заболевание, которое возникает в результате первичной недостаточности нейраминидазы из-за мутаций в гене (NEU1), кодирующем этот белок, расположенном на хромосоме 6p21.33. Галактосиалидоз вызывается дефицитом двух лизосомальных ферментов — нейраминидазы и β-галактозидазы. Утрата активности этих ферментов является результатом мутаций в одном гене, CTSA, расположенном на хромосоме 20q13.12, кодирующей протективный белок катепсин А, функция которого — обеспечение стабильности лизосомальных ферментов и защита их от деградации.
Нейраминидаза расщепляет концевые сиалильные связи нескольких олигосахаридов и гликопротеинов. Недостаточность этого фермента приводит к накоплению олигосахаридов и экскреции с мочой сиализированных концевых олигосахаридов и сиалилгликопептидов. Исследование тканей заболевших выявляет патологическое накопление субстрата во многих тканях, включая печень, костный мозг и ГМ.
Клинические фенотипы, связанные с дефицитом нейраминидазы, разнообразны. Для сиалидоза типа I, который обычно проявляется во втором десятилетии жизни, характерны миоклония и симптом «вишневой косточки» в макуле. У таких пациентов обычно возникают вторичные симптомы, такие как нарушения походки, миоклонуса или нарушения зрения. Напротив, сиалидоз типа II может возникнуть в разном возрасте (врожденный, младенческий и ювенильный), в зависимости от тяжести мутации гена.
Врожденная и младенческая формы являются результатом изолированного дефицита нейраминидазы, тогда как ювенильная форма — результатом недостаточности как нейраминидазы, так и β-галактозидазы. Врожденное заболевание типа II характеризуется водянкой плода, асцитом новорожденных, гепатоспленомегалией, нарушением структуры эпифизов, уплотнением надкостницы, а также мертворождением или смертью в периоде новорожденности. Инфантильная форма типа II проявляется в первый год жизни множественным дизостозом, умеренной задержкой психического развития, висцеромегалией, помутнением роговицы, симптомом «вишневой косточки» и судорогами.
Ювенильную форму сиалидоза типа II называют галактосиалидозом. Она может проявляться в разном возрасте — от грудного до взрослого.
Клинические проявления у детей первого года жизни схожи с фенотипом ганглиозидоза GM1 и проявляются в виде отеков, асцита, дисплазией костей и симптомом «вишневой косточки».
У пациентов с более поздним началом заболевания наблюдаются множественный дизостоз, висцеромегалия, умственная отсталость, дисморфизм, помутнение роговицы, прогрессирующие неврологические нарушения и симптом «вишневой косточки».
Для любой формы заболевания не существует специфической терапии, хотя исследования на животных продемонстрировали улучшение клинической картины после трансплантации костного мозга. Диагноз сиалидоза и галактосиалидоза устанавливается на основании снижения активности соответствующих ферментов и выявления мутаций в ответственном гене. Определение активности этих ферментов в культуре амниоцитов и ворсинках хориона и/или выявление определенных генных мутаций позволяет проводить пренатальную диагностику.
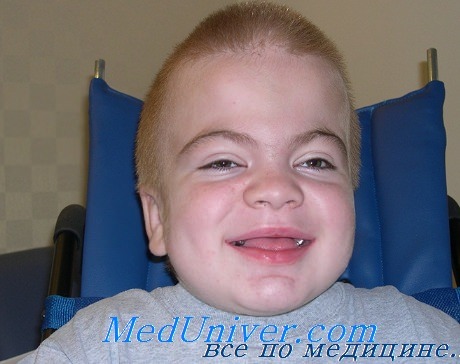
Сиалидоз у ребенка
б) Аспартилглюкозаминурия. Это АуР-лизосомная болезнь накопления встречается редко, за исключением Финляндии, где частота носителей оценивается у 1 из 36 взрослых. Высокая частота обусловлена геном-основателем. Нарушение является результатом недостаточности аспартилглюкозаминидазы, что приводит к накоплению аспартилглюкозамина, особенно в печени, селезенке и ЩЖ.
Ген фермента (AGA) находится на хромосоме 4q32-33. ДНК и ген были выделены и секвенированы. В Финляндии большинство заболеваний связано с единственной мутацией AGA, кодирующейся p.C163S, в то время как среди др. популяций выявлено множество разных мутаций.
Симптомы аспартилглюкозаминурии обычно проявляются на первом году жизни в виде рецидивирующих инфекций, диарей и пупочных грыж. Позднее отмечается огрубление черт лица и низкорослость. Др. признаки включают слабость суставов, макроглоссию, хриплый голос, помутнение хрусталика, гипотонию и спастичность. Психомоторное развитие обычно начинает отставать от нормы после 5-летнего возраста.
Часто наблюдаются отклонения в поведении, а значения IQ у взрослых с этим заболеванием обычно <40 (тяжелая умственная отсталость). Больные, как правило, доживают до зрелого возрасте, а ранняя смерть связана нередко с пневмонией или др. легочной патологией. Окончательный диагноз требует определения активности фермента аспартилглюкозаминидазы в лейкоцитах периферической крови и/или выявления специфической мутации(й) AGA.
Несколько пациентов перенесли аллогенную трансплантацию костного мозга, но данный подход в лечении оказался неэффективным, поэтому специального лечения не существует. Пренатальная диагностика доступна при определении недостаточности аспартилглюкозаминидазы и/или специфических мутаций AGA в культуре амниоцитов или ворсинах хориона.
в) α-Маннозидоз. Данное АуР-заболевание возникает в результате недостаточности α-маннозидазы и накопления соединений, богатых маннозой. Ген MAN2B1, кодирующий фермент, находится на хромосоме 19p13.2-q12, также были определены кДНК и последовательность гена. На сегодняшний день зарегистрировано >140 генных мутаций. Болезнь может иметь гетерогенную клиническую картину. Выделяют тяжелую младенческую форму, или болезнь типа I, и более мягкий ювенильный вариант, болезнь типа II.
У всех пациентов наблюдаются задержка психомоторного развития, грубые черты лица и множественный дизостоз. Однако младенческая форма характеризуется более быстрым ухудшением психических функций и смертью в возрасте от 3 до 10 лет. Пациенты с младенческой формой также имеют более тяжелое поражение скелета и ге-патоспленомегалию. Ювенильное расстройство характеризуется появлением симптомов в раннем детстве или подростковом возрасте с более легкими соматическими признаками. Пациенты, как правило, доживают до взрослого возраста.
У пациентов с типом II наблюдались потеря слуха, деструктивный синовит, панцитопеня и спастическая параплегия. Диагноз ставится на основании определения активности а-маннозидазы в лейкоцитах или культуре фибробластов. Клинические испытания ФЗТ с рекомбинантной человеческой α-маннозидазой продолжаются. Пренатальный диагноз м.б. поставлен путем определения дефекта фермента и/или конкретных генных мутаций в культуре амниоцитов или ворсинах хориона.