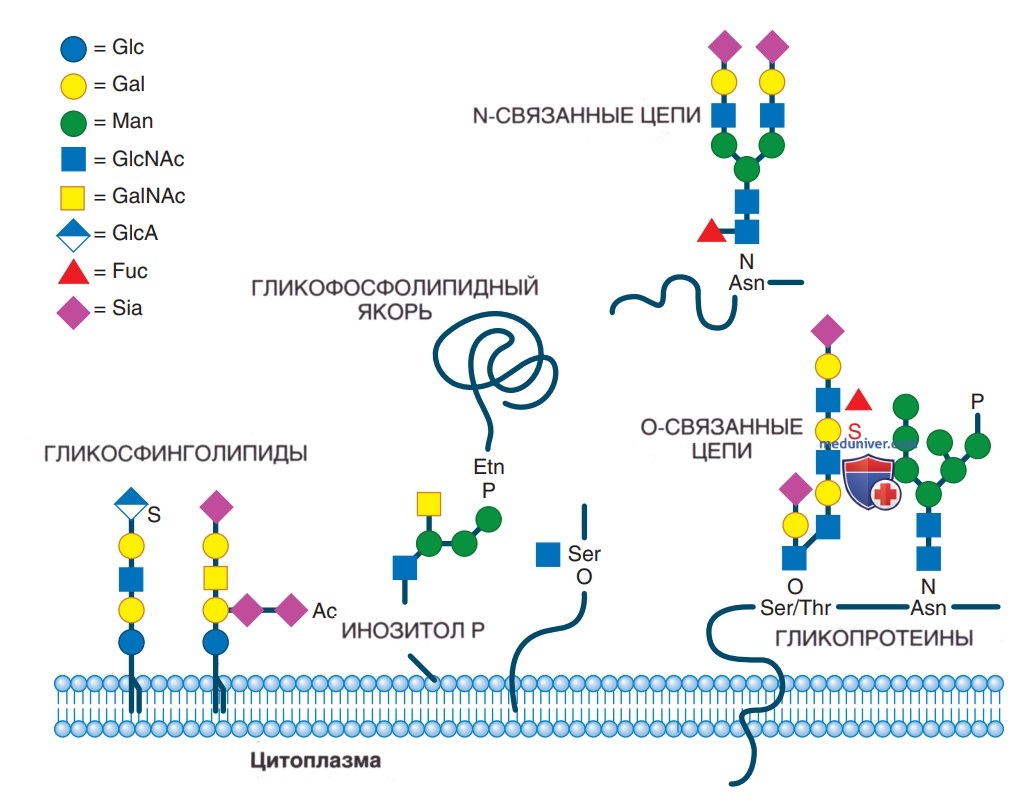
Гликозилирование является сложным многоступенчатым метаболическим процессом добавления (олиго) сахаридов к белкам и липидам. Классификация нарушений гипогликозилирования основана на биохимическом типе дефекта гликозилирования: 1) дефекты N-гликозилирования, 2) дефекты О-гликозилирования, 3) дефекты гликозилирования липидов и гликозилирования гликозилфосфадил-инозитола, 4) дефекты в других путях гликозилирования и множественные дефекты (рис. 1). Нет известных нарушений, возникающих в результате аномального С-связанного гликозилирования. CDG маркируются на основании их генетического дефекта.
Рисунок 1. Схема различных типов гликозилирования. Слева направо: гликосфинголипиды, гликофосфолипидный якорь (GPI-якорь), О-гликозилирование мембранного белка, N-гликозилирование мембраны и секреторный N-гликан
Важным путем является гликозилирование белков. Большинство функциональных белков гликозилированы, включая белки сыворотки крови [напр., трансферрин, церулоплазмин, тироксинсвязывающий глобулин (ТСГ)], гормоны (напр., ТТГ, ФСГ, ФГ, АКТГ, ИФРСБ), а также факторы свертывания крови и антикоагуляции (напр., факторы IX и XI, антитромбин). Мембранные белки также сильно гликозилированы. Важные в/клеточные гликопротеины включают ферменты, такие как гликозилтрансферазы или лизосомальные ферменты.
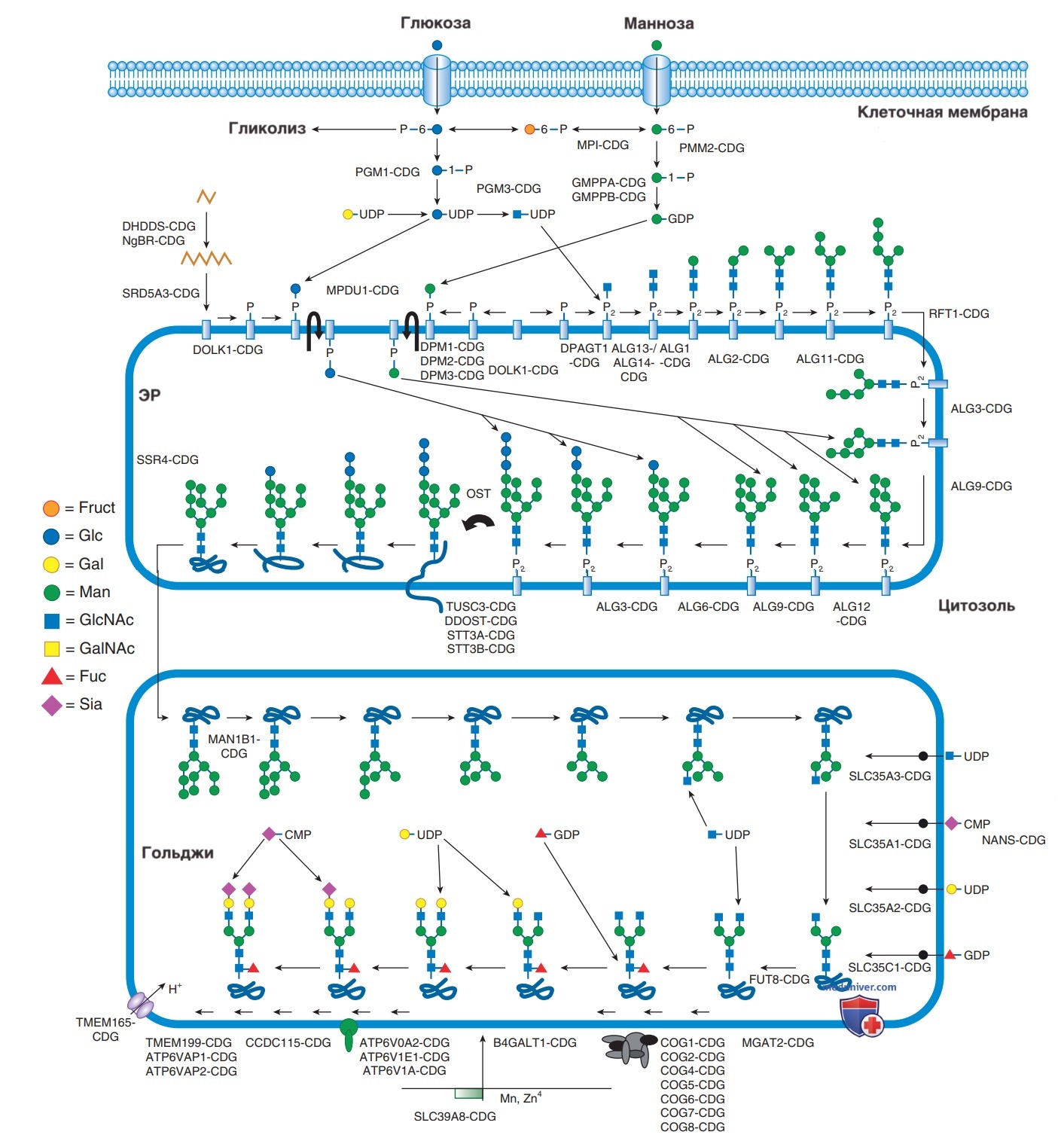
N-гликаны связаны с амидной группой аспарагина. Синтез N-гликанов представляет собой сложный процесс и начинается с образования нуклеотидсвязанных сахаров, сборки (в цитозоле и эндоплазматическом ретикулуме) и обработки (в аппарате Гольджи) (рис. 2). Большинство заболеваний у детей представляют собой нарушения N-гликозилирования. О-гликаны связаны с гидроксильной группой серина или треонина. Синтез О-гликанов происходит преимущественно в комплексе Гольджи; их дефекты могут включать ксилозилирование, фукозилирование, маннозилирование или др. модификации. Большое внимание уделяется дефектам О-маннозилирования из-за их значимости при дистрогликанопатиях.
Рисунок 2. Обзор разл. отделов клетки, участвующих в N-гликозилировании. Активация нуклеотидных сахаров в цитоплазме сопровождается многостадийным синтезом гликанов, связанным с долихолом в эндоплазматическом ретикулуме. Трансфер гликана от липидного плеча к белку сопровождается транспортом к комплексу Гольджи для дальнейших модификаций. ЭР — эндоплазматический ретикулум
Гликозилирование липидов — важный процесс для синтеза церамидов и ганглиозидов. Гликозилфосфатидилинозитолы (GPI) являются особыми гликолипидами, связывающими разл. белки с плазматической мембраной в качестве сложных липидно-сахарных якорей (GPI-якоря, см. рис. 1).
CDG — это преимущественно мультисистемные заболевания, вызванные >140 разл. генетическими дефектами в синтезе гликопротеина и гликолипидных гликанов. Эта группа нарушений является новой и быстрорастущей и, возможно, будет наиболее крупной среди других нарушений обменных процессов. Большинство пациентов с CDG имеют дефекты N-гликозилирования, за ними следует самая быстрорастущая группа CDG, включающая множественные пути гликозилирования и синтез долихолфосфата. Меньшие группы представляют собой нарушения О-гликозилирования и нарушения гликозилфосфатидилинозита.
Самое «старое» CDG — это PMM2-CDG, генетический дефект которого приводит к потере фосфоманномутазы-2 (ФММ2), фермента, который катализирует превращение маннозо-6-фосфата в маннозо-1-фосфат. Большинство CDG имеют АуР-наследование. Только два N-связанных CDG являются АуД: GANAB-CDG и PRKCSH-CDG. Доминантно наследуемые О-связанные CDG включают EXT1/EXT2-CDG, POFUT1-CDG и POGLUT1-CDG. Х-связанные ВНЕ включают ALG13-CDG, SSR4-CDG, PIGA-CDG, SLC35A2-CDG, АТР6АР2-CDG и ATP6AP1-CDG.
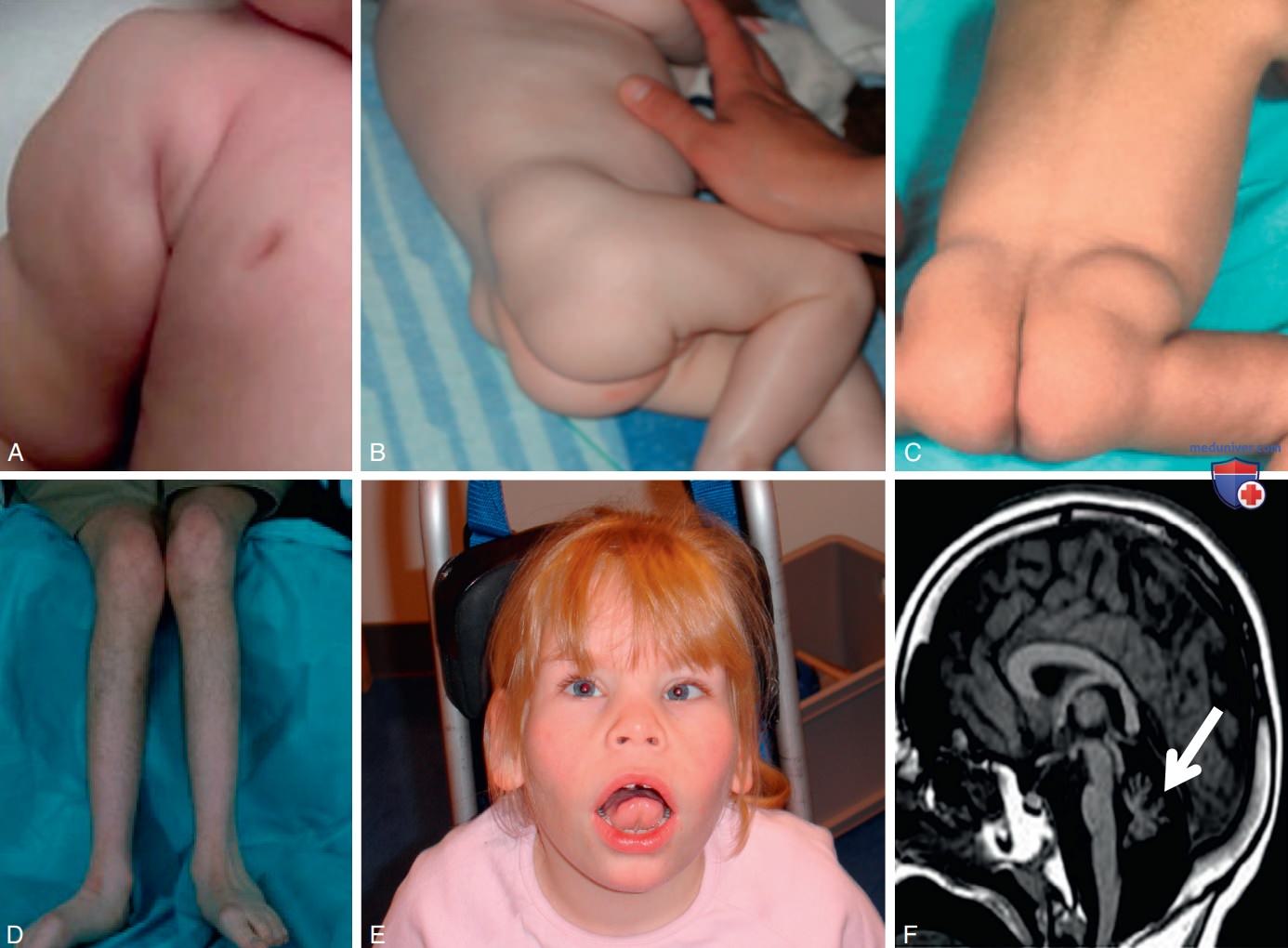
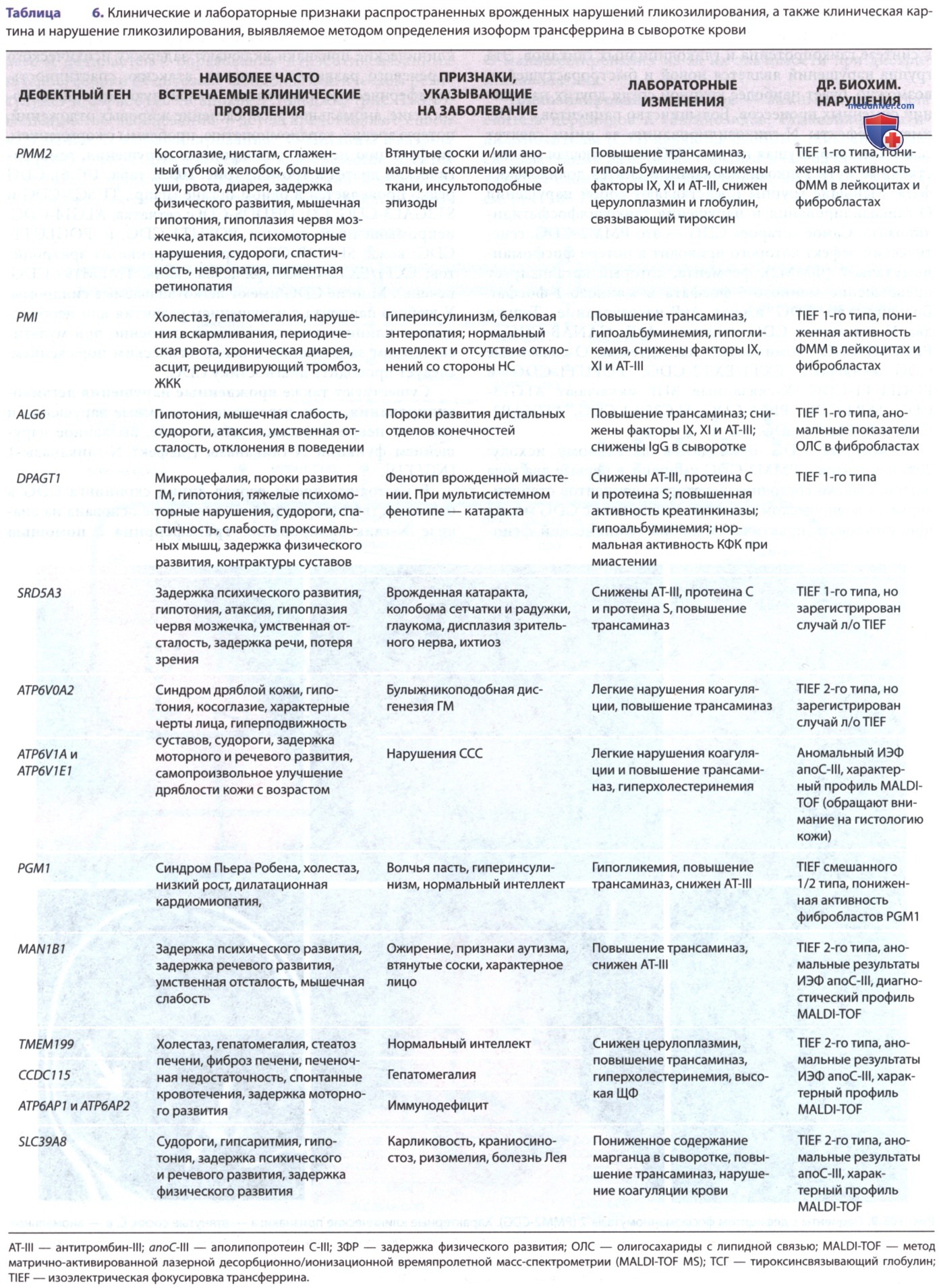
Некоторые CDG приводят к летальному исходу: 20% пациентов с PMM2-CDG умирают в первые два года жизни. Однако состояние некоторых пациентов стабилизируется в юношеском возрасте. У пациента с CDG может присутствовать практически любой клинический фенотип. Заболевание может затронуть любой орган или систему органов и чаще всего влияет на ЦНС. Наиболее частые клинические признаки включают задержку психического и речевого развития, судороги, атаксию, спастичность, периферическую невропатию, мышечную гипотонию, косоглазие, аномальное распределение жировых отложений, потерю зрения, кардиомиопатию, проблемы с кормлением, дисфункцию печени, эндокринные нарушения, геморрагический диатез и тромбоз (рис. 3, табл. 6).
Рисунок 3. Пациенты с дефицитом фосфоманномутазы-2 (PMM2-CDG). Характерные клинические признаки: А — втянутые соски; B, C — аномальное распределение жировых отложений; D — мышечная атрофия на фоне периферической невропатии, возникшей после пубертата; E — характерные черты лица: страбизм, короткий нос, вывернутые вперед ноздри, длинный подносовой желобок и большие уши; F — на магнитно-резонансной томографии головного мозга (ГМ) в Т1-взвешенном режиме в сагиттальной проекции наблюдается гипоплазия червя мозжечка (стрелка) и атрофия ГМ
CDG редко проявляется в одном органе (напр., TUSC3-CDG и ST3GAL3-CDG: ГМ; DHDDS-CDG: сетчатка; ALG14-CDG: нейромышечный синапс; POFUT1-CDG и POGLUT1-CDG: кожа; SEC23B-CDG: происхождение из эритроцитов; EXT1/EXT2-CDG: хрящевая ткань; TMEM199-CDG: печень). Многие CDG имеют легкоузнаваемые синдромы. У любого пациента с нарушением развития или необъяснимым клиническим состоянием, особенно при мульти-системном заболевании с неврологическим поражением, следует проводить диагностику CDG.
Существуют также врожденные нарушения дегликозилирования, в т.ч. известные лизосомные нарушения и тяжелое неврологическое заболевание, вызванное нарушением функции N-гликаназы ([дефект N-гликаназы-1 (NGLY1)].
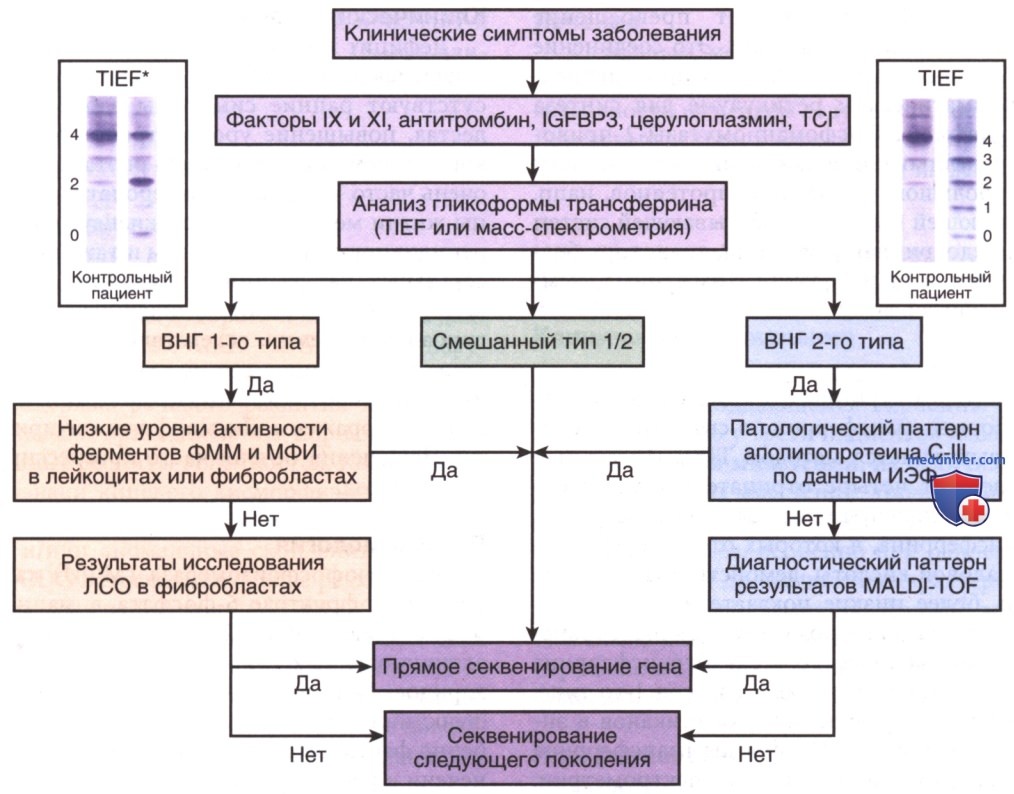
В настоящее время первая линия скрининга CDG в большинстве лабораторий во всем мире основана на анализе N-гликозилирования трансферрина с помощью изоэлектрической фокусировки (TIEF). Этот метод позволяет проводить разделение изоформ трансферрина на основе состояния заряда концевых остатков сиаловой кислоты. Гипосиалилированные изоформы трансферрина (у которых отсутствуют концевые остатки сиаловой кислоты) дают разл. катодные сдвиги в зависимости от недостающих или усеченных гликановых цепочек.
Паттерн 1-го типа указывает на ранний метаболический дефект в синтезе и сборке гликанов, связанных с цитозолем эндоплазматического ретикулума. Паттерн 2-го типа указывает на дефекты продуцирования гликанов, связанные с комплексом Гольджи (рис. 4).
Рисунок 4. Диагностическая блок-схема нарушений гликозилирования, влияющих на N-гликозилирование. *Вместо TIEF можно использовать методы масс-спектрометрии; IGFBP3 — белок 3, связывающий инсулиноподобный фактор роста; ТСГ — тироксинсвязывающий глобулин; CDG — врожденные нарушения гликозилирования; ФММ — фосфоманномутаза; МФИ — маннозофосфоизомераза; ЛСО — липидно-связанные олигосахариды; TIEF — изоэлектрическая фокусировка трансферрина; ИЭФ — изоэлектрическая фокусировка; MALDI-TOF — метод матрично-активированной лазерной десорбционно/ионизационной времяпролетной масс-спектрометрии
Изоэлектрическая фокусировка аполипопротеина С-III (ИЭФ апоС-III), сывороточного О-гликозилированного белка позволяет выявить некоторые нарушения О-гликозилирования (сочетание дефектов N- и О-гликозилирования). Также для подтверждения CDG1, смешанных профилей CDG I/II и для дальнейшего подтипирования дефектов CDG1 можно использовать масс-спектрометрию с высоким разрешением интактного трансферрина. При определенных типах CDG (преимущественно при паттерне 2-го типа, связанного с комплексом Гольджи) м.б. использована гликомика, полученная методом матричноактивированной лазерной десорбционно/ионизационной времяпролетной масс-спектрометрии (MALDI-TOFMS).
Анализ долихол-связанного гликана или липид-связанного олигосахарида является сложным, но чувствительным методом обнаружения дефектов сборки N-гликана (CDG 1-го типа), связанных с эндоплазматическим ретикулумом, в фибробластах пациента. Рецидивирующее повышение уровня ЩФ в крови может указывать на дефекты GPI-якоря.
Дистрогликанопатии м.б. подтверждены при аномальных результатах иммуногистохим. исследований биоптатов мышц. Анализ прикрепленных к мембране маркеров CD16 и CD24 в лейкоцитах методом сортировки клеток с активированной флуоресценцией (СКАФ) с высокой вероятностью указывает на аномалию GPI-якоря, особенно при значительном повышении ЩФ в крови. Ферментный анализ крови выполняют только для нескольких, наиболее распространенных CDG [PMM2-CDG, маннозо-6-фосфат-изомераза (MPI; англ. Mannose-6 phosphate isomerase) - CDG, ФММ1-ВНГ]; он дает более надежные результаты при анализе фибробластов.
При отклонениях паттерна TIEF или клиническом подозрении на любой тип CDG большинство медицинских центров используют прямой анализ генной панели CDG или секвенирование следующего поколения (полное секвенирование экзома) (см. рис. 4).
а) Врожденные нарушения N-гликозилирования:
1. Недостаточность фосфоманномутазы-2 (PMM2-CDG):
- Клинические проявления. PMM2-CDG — самый распространенный и легко узнаваемый CDG. У большинства пациентов уже в первые месяцы жизни наблюдаются перемежающееся косоглазие, характерные черты лица (короткий нос, длинный подносовый желобок, большие уши) (см. рис. 3), втянутые соски и/или аномальные скопления жировой ткани (рис. 3), проблемы с кормлением, мышечная гипотония и сниженные рефлексы. Также часто наблюдается нистагм (вызванный понтоцеребеллярной гипоплазией и гипоплазией мозжечка; см. рис. 3). У большинства пациентов присутствует задержка психомоторного развития, но у некоторых больных было описано нормальное интеллектуальное развитие. У большинства пациентов развивается заболевание, характеризующееся мультисистемным поражением.
Однако у <25% больных наблюдаются изолированный неврологический фенотип без вовлечения других органов, нормальная эндокринная регуляция и отсутствие коагулопатии. Проявления со стороны НС довольно разнообразны. К наиболее распространенным относятся атаксия, судороги, спастичность и периферическая невропатия (см. рис. 3). Кроме того, могут возникать дистония, инсультоподобные эпизоды и проксимальная миопатия.
PMM2-CDG не относится к прогрессирующим заболеваниям, однако определенные признаки могут проявляться в разные возрастные периоды. С рождения могут наблюдаться скопления жидкости в перикарде, кардиомиопатия или расстройства ЖКТ в виде хронической рвоты/диареи. После семи лет развиваются пигментная ретинопатия и катаракта, в пубертатном периоде — сколиоз, невропатия и рецидивирующие тромбозы. Функция печени нарушается незначительно, и только у некоторых пациентов развивается холестаз или фиброз печени. У большинства пациентов наблюдается гипергонадотропный гипогонадизм; случаев успешных беременностей не зарегистрировано. Умственная отсталость м.б. как легкой, так и тяжелой степени. Развитие речи часто происходит с задержкой и даже может отсутствовать.
Часто наблюдается аутичное поведение, хотя пациенты обладают жизнерадостным характером.
- Патофизиология. Фосфоманномутаза-2 катализирует превращение маннозо-6-фосфата в маннозо-1-фосфат. Это соединение является донором маннозных единиц, которые используются в эндоплазматическом ретикулуме для синтеза олигосахаридов. Дефицит фосфоманномутазы-2 приводит к нарушению гликозилирования, недостаточности или дисфункции большого числа гликопротеинов, напр. факторов свертывающей и противосвертывающей систем крови. Страдает эндокринная регуляция, транспорт белков, функция печени, а также функция иммунных, мембранных и рецепторных белков.
- Диагностика. Первичным методом скрининга PMM2-CDG является анализ гликоформы трансферрина в сыворотке, который чаще всего выполняется методом TIEE Интактный трансферрин содержит четыре отрицательно заряженных остатка сиаловой кислоты (тетрасиалотрансферрин). Гликоформы трансферрина, в которых отсутствуют концевые остатки сиаловой кислоты, демонстрируют разные катодные сдвиги, более низкие показатели тетрасиалотрансферрина, более высокие показатели дисиалотрансферрина и некоторое количество α-сиалотрансферрина (см. рис. 4). Это так называемый паттерн 1-го типа. Он свидетельствует о нарушении сборки гликанов в эндоплазматическом ретикулуме.
Изоформы трансферрина можно также обнаружить методом масс-спектрометрии. К формированию л/п-паттерна изоформы трансферрина могут приводить др. нарушения, в т.ч. галактоземия, наследственная непереносимость фруктозы и злоупотребление алкоголем. Анализ ферментов ФММ м.б. проведен в лейкоцитах и фибробластах.
Высокие уровни трансаминаз сыворотки, гипоальбуминемия, низкие уровни активности факторов IX и XI, а также антитромбина, или низкие уровни церулоплазмина, или тироксинсвязывающего глобулина с высокой степенью вероятности указывают на CDG, в том числе на самый распространенный тип — PMM2-CDG.
PMM2-CDG наследуется АуР. Генетическое исследование, как правило, выполняют методом прямого секвенирования. Наиболее распространенный патогенетический вариант (c.422G>A; R141H) обнаруживают у 75% пациентов европеоидной расы. Точная частота встречаемости PMM2-CDG неизвестна, но по оценкам она составляет до 1:40 000-80 000 человек в Европе. Пренатальный диагноз можно достоверно установить только при проведении генетических исследований.
- Лечение. Основным лечением PMM2-CDG является симптоматическая терапия. Даже при наилучшем лечении смертность в первые 2 года жизни составляет около 20%, летальный исход наступает в основном от осложнений на сердце или почки, а также от тяжелых инфекций. В настоящее время рекомендованная терапия включает диету, при необходимости зондовое питание, поддержку сердечной деятельности, гормональную терапию, физиотерапию и реабилитационную терапию, логопедические занятия, лечение судорог и хирургическое вмешательство при косоглазии. К разрабатываемым терапиям относятся таргетное лечение маннозофосфатом и лечение ферментными заместителями. Эти разработки находятся только на этапах доклинических испытаний.
2. Дефицит маннозофосфоизомеразы (MPI-CDG):
- Клинические проявления. Дефицит MPI является клинически узнаваемой и поддающейся лечению CDG. У большинства пациентов присутствуют ранние симптомы заболевания печени (холестаз, повышение уровня трансаминаз) и трудности с кормлением, повторяющаяся рвота, хроническая диарея и очень часто — белковая энтеропатия. Уже в первые месяцы жизни могут появиться жизнеугрожающие эпизоды с рецидивирующим тромбозом и тяжелым ЖКК вследствие серьезных нарушений коагуляции. Причиной гипогликемии обычно является гиперинсулинизм. М.б. тяжелая гипоальбуминемия; вследствие сочетания асцита и гепатомегалии у пациентов может наблюдаться видимое увеличение живота. У пациентов с MPI-CDG др. органы и ЦНС не поражены. Дисморфические признаки отсутствуют. Нарушения печени часто прогрессируют до фиброза или цирроза.
- Патофизиология. Маннозофосфоизомераза (МФИ) катализирует превращение фруктозо-6-фосфата в маннозо-6-фосфат за одну стадию до ФММ2, тем самым блокируя образование активированных единиц маннозы для синтеза олигосахаридов. Гипогликозилирование приводит к нарушению функции гликопротеинов, как и в случае ФММ2-ВНГ, особенно факторов свертывания и антикоагуляции, функции печени и рецепторов гормонов.
- Диагностика. Основным методом скрининга у пациентов с подозрением на MPI-CDG является анализ изоформ трансферрина в сыворотке методом ИЭФТ (см. рис. 4) или масс-спектрометрии. Дефицит МФИ приводит к паттерну 1-го типа, что очевидно при дефиците РММ2. Анализ ферментов МФИ м.б. проведен в лейкоцитах и фибробластах. Повышенное содержание трансаминаз в сыворотке, гипоальбуминемия, пониженная активность факторов IX и XI и антитромбина, гиперинсулинизм и некетотическая гипогликемия с высокой вероятностью указывают на наличие MPI-CDG.
MPI-CDG наследуется АуР. Генетическое тестирование обычно выполняется путем прямого секвенирования. Точные показатели заболеваемости MPI-CDG неизвестны, однако в Европе они, по оценкам, составляют 1:800 000. Пренатальный диагноз можно достоверно установить только при проведении генетических исследований. Хотя данная форма CDG встречается редко, ранняя диагностика имеет решающее значение, поскольку это заболевание поддается лечению.
- Лечение. MPI-CDG — первый из всех типов CDG, который начали лечить методом диетотерапии. Манноза оказывает клинический эффект как при в/в, так и при пероральном применении 1 г/кг в сутки в 3-4 приема (в РФ была предложена дозировка 200 мг/кг 4-6 р/сут с целью поддержания уровня маннозы в сыворотке, достаточного для снижения эндокринных нарушений, дефектов коагуляции и энтеропатии)*. Известным побочным эффектом терапии маннозой является гемолиз. Этот метод терапии основан на использовании альтернативного пути: манноза может фосфорилироваться гексокиназой до маннозо-6-фосфата, обходя дефект МФИ. Клиническая симптоматика улучшается быстро, однако функции печени могут и дальше ухудшаться. В связи с фиброзом и циррозом печени может потребоваться трансплантация, которая приводит к разрешению метаболического заболевания. Самая длительная продолжительность жизни пациента с MPI-CDG составила >35 лет.
P.S. * Иванов Д.О., Новикова В.П., Похлебкина А.А. Врожденные нарушения гликозилирования // Педиатрия. 2018. Т. 9. № 3. С. 12.
- Клинические проявления. ALG6-CDG — второй по распространенности тип CDG. У большинства пациентов отмечается мышечная гипотония, мышечная слабость, судороги и атаксия. На данный момент у всех пациентов с ALG6-CDG имеются нарушения интеллекта. Часто отмечают такие неврологические симптомы, как задержка речевого развития и нистагм. У пациентов наблюдают брахидактилию, пороки развития скелета и поперечные дефекты конечностей. Косоглазие и характерные признаки лицевого дисморфизма (гипертелоризм, овальное лицо, короткий нос) встречаются редко. Втянутые соски и/или аномальные скопления жировой ткани при ALG6-CDG встречаются крайне редко.
У наиболее тяжелых пациентов с ALG6-CDG в первые месяцы жизни появляются симптомы мультисистемных поражений: тяжелые инфекции, белковая энтеропатия, гипоальбуминемия, анемия и отсутствие весовых прибавок. У нескольких пациентов наблюдали аутистическое поведение и изменения настроения. Самому старшему пациенту на данный момент почти 45 лет.
- Патофизиология. Метаболический дефект обусловлен нарушением связывания первой из трех глюкоз с липоолигосахаридом в эндоплазматическом ретикулуме. Это связывание глюкозы играет очень важную роль в прикреплении ферментного комплекса олигосахарилтрансферазы к вновь образованной олигосахаридной цепи и в способности переносить ее к белку. Это приводит к гипогликозилированию белка и нарушению функции гликопротеина, сходному с нарушениями при PMM2-CDG и MPI-CDG. Изменения лабораторных показателей, как и при ранее описанных CDG, включают снижение факторов свертывающей и антисвертывающей систем крови, уровня гормонов ЩЖ и IgG, нарушения функции печени.
- Диагностика. Основным методом скрининга пациентов с подозрением на ALG6-CDG является анализ гликозилирования трансферрина с помощью изоэлектрической фокусировки (TIEF) или методом масс-спектрометрии. Дефицит ALG6 приводит к развитию паттерна, характерного для 1-го типа (см. рис. 4), который наблюдается при дефиците ФММ2 и МФИ. Методов ферментного анализа не существует, однако можно оценить уровни липоолигосахаридов в фибробластах пациента.
ALG6-CDG наследуется АуР. Генетическое тестирование обычно выполняется путем прямого секвенирования. Наиболее распространенными мутациями являются P-A333V и p.I299Del. Пренатальный диагноз можно достоверно установить только при проведении генетических исследований. Точные показатели распространенности ALG6-CDG неизвестны.
- Лечение. Основным методом лечения ALG6-CDG на данный момент является симптоматическая терапия. Показатели летальности на первом году жизни составляют около 10%. Летальные исходы в большинстве случаев связаны с белковой энтеропатией и тяжелыми инфекциями.
- Клинические проявления. Дефицит DPAGT1 является распознаваемой и потенциально поддающейся лечению формой CDG. Приблизительно у трети пациентов наблюдаются симптомы врожденной миастении, не отличимые от других видов генетических миастений. Уровни КФК не отклоняются от нормы. У таких пациентов отмечается относительно неплохой прогноз, особенно при раннем начале лечения миастении. Для др. пациентов характерен мультисистемный тип поражения с микроцефалией, пороками развития ГМ, мышечной гипотонией, тяжелыми психомоторными нарушениями, судорогами, спастичностью, отсутствием прибавки МТ, контрактурами суставов и катарактой.
- Патофизиология. Дефект DPAGT1 приводит к замедлению реакции присоединения второго сахара GlcNAc к фосфорилированному плечу долихола. В результате синтез гликанов за пределами мембраны эндоплазматического ретикулума прерывается на очень раннем этапе. Нарушение гликозилирования рецептора в нервно-мышечных синапсах приводит к развитию миастении. Гипогликозилирование при мультисистемном типе приводит к нарушению функции гликопротеина, сходному с тем, которое наблюдается при PMM2-CDG. В частности, при мультисистемном типе также вовлекаются факторы коагуляции. Что интересно, указанные изменения приводят к высоким уровням КФК (в отличие от типа врожденной миастении) и гипоальбуминемии.
- Диагностика. Основным методом скрининга является анализ гликозилирования трансферрина с помощью TIEF или методом масс-спектрометрии. У большинства пациентов наблюдается паттерн, характерный для 1-го типа (см. рис. 4), однако у пациентов с типом врожденной миастении результаты скрининга м.б. нормальными. Клинически доступный анализ на ферменты отсутствует.
DPAGT1-CDG наследуется АуР. Генетическое тестирование обычно выполняется путем прямого секвенирования. Точная заболеваемость неизвестна. Пренатальный диагноз можно достоверно установить только при проведении генетических исследований. Поскольку у нескольких пациентов с миастеническим фенотипом были получены л/о результаты TIEF, в подозрительных случаях, особенно в целях определения возможных схем терапии, предлагается использовать панель тестов на врожденную миастению.
- Лечение. Фенотип врожденной миастении нередко поддается лечению высокими дозами пиридостигмина. В конечном счете симптомы улучшаются на фоне применения сальбутамола. Лечение мультисистемного типа DPAGT1-CDG симптоматическое.
б) Врожденные дефекты О-гликозилирования:
1. Синдром окулоцеребральной дисплазии-мышечной дистрофии и миоокулоцеребральный синдром (POMT1-CDG, POMT2-CDG, POMGNT1-CDG). Данная группа дефектов О-гликозилирования, от изолированной мышечной дистрофии до синдрома Уокера-Варбурга, проявляется выраженной мышечной слабостью, врожденными пороками развития глаз и нарушением миграции нейронов.
Могут наблюдаться пахигирия, булыжноподобная дисгенезия ГМ, гидроцефалия, полимикрогирия, гетеротопия и агенезия мозолистого тела. К порокам развития глаз относятся анофтальмия, микрофтальмия, врожденная катаракта или колобомы. Врожденная мышечная дистрофия сопровождается значительным повышением уровня КФК. Отмечают тяжелые психомоторные нарушения.
В основе заболевания лежит нарушение синтеза О-маннозилгликанового ядра, которое играет важную роль в правильном гликозилировании α-дистрогликана. α-дистрогликан подвергается интенсивному О-гликозилированию остатками маннозы и экспрессируется в мышцах и головном мозге. Нарушение маннозилирования α-дистрогликана приводит к дегенерации мышц и нарушениям миграции. При иммуногистохим. исследовании биоптатов мышц обнаруживают патологическое окрашивание α-дистрогликана.
У пациентов с изолированными нарушениями О-маннозилирования результаты изоэлектрической фокусировки трансферрина не отклоняются от нормы. Клинически доступного анализа ферментов не существует. Диагноз устанавливают на основании результатов гист. исследования (биоптатов мышц) и генетического анализа.
POMT1-CDG, POMT2-CDG, POMGNT1-CDG являются наиболее распространенными АуР-α-дистрогликанопатиями. Отмечаются и дефекты др. генов, кодирующих ферменты этого пути гликозилирования. При заболевании у человека описаны дефекты следующих генов: РОМК, FKTN, FKRP, LARGE, B4GAT1, ТМЕМ5 и ISPD. Точные показатели распространенности а-дистрогликанопатий неизвестны.
Лечение α-дистрогликанопатий является симптоматическим.
в) Дефекты в гликозилировании липидов и биосинтезе гликозилфосфатидил-инозитол-якоря:
1. Синдром гиперфосфатазии — умственной отсталости: дефицит PIGA (PIGA-CDG). Клиническая картина этого синдрома включает синдром эпилепсии с умственной отсталостью, гипотонией, дисморфическими чертами лица, аномалиями кожи, врожденными пороками развития ГМ и поведенческими аномалиями, в т.ч. РАС. Зарегистрированы также случаи др. пороков развития органов, в т.ч. нарушения работы сердца и почек. (Важно отметить, что соматические мутации с дефектом PIGA также могут приводить к пароксизмальной ночной гемоглобинурии.)
GlcNAc не может эффективно транспортироваться на фосфатидилинозитол для синтеза гликофосфатидилинозита. Аномальное прикрепление ЩФ приводит к гиперфосфатаземии в крови и потере специфических поверхностных АГН в клетках крови.
При дефектах GPI-якоря анализ изоформы трансферрина соответствует норме. СФАК-анализ прикрепленных к мембране маркеров CD16 и CD24 в лейкоцитах в значительной степени указывает на аномалию GPI-якоря, особенно в сочетании с повышенным содержанием ЩФ в сыворотке. Анализ мутаций подтверждает дефект.
PIGA-CDG является Х-сцепленным. Точная заболеваемость неизвестна. Похожий фенотип был описан при дефектах PIGO, PIGV, PIGY, PIG, PGAP2 и PGAP3.
Лечение PIGA-CDG симптоматическое.
г) Дефекты в других путях гликозилирования и множественные дефекты, в том числе дефекты биосинтеза долихолфосфатов:
1. Дефицит стероид-5а-редуктазы (SRD5A3-CDG):
- Клинические проявления. Дефицит стероид-5α-редуктазы (SRD5A3) представляет собой клинически распознаваемый CDG, первоначально описанный как синдром множественных врожденных пороков развития. Диагноз был поставлен примерно двадцати пациентам разного возраста, в т.ч. одному пациенту в 45 лет. У пациентов наблюдается гипотония, атаксия и аномалии зрения, в т.ч. врожденная катаракта, колобомы сетчатки и радужной оболочки глаза, глаукома, дисплазия зрительного нерва и потеря зрения. Гипоплазия червя мозжечка м.б. вариабельной. У всех пациентов с этой патологией, зарегистрированных в настоящее время, наблюдалась умственная отсталость. Около трети пациентов страдают тяжелым врожденным ихтиозом. Часто встречаются гипертрихоз и дисморфические черты лица, в том числе квадратное лицо, высокий лоб, большие уши и грубые черты лица.
У некоторых детей с SRD5A3-CDG наблюдаются тяжелые нарушения аутистического спектра. Деформации скелета (сколиоз) и пороки сердца встречаются реже.
- Патофизиология. Дефицит SRD5A3 приводит к аномальному синтезу долихола, влияющему на ранний синтез гликанов вне мембраны ЭР, а также влияет на О-маннозилирование и синтез GPI-якоря. Гипогликозилирование влияет на антикоагулянтные факторы свертывания крови и приводит к повышению трансаминаз.
- Диагностика. Основным методом скрининга для пациентов с подозрением на SRD5A3-CDG является анализ гликозилирования трансферрина с помощью TIEF или методом масс-спектрометрии. У большинства пациентов наблюдается паттерн 1-го типа (см. рис. 4), но было описано несколько л/о случаев. Клинически доступного ферментного анализа не существует.
SRD5A3-CDG наследуется АуР. Генетическое тестирование обычно выполняется путем прямого секвенирования. Точная заболеваемость неизвестна.
- Лечение. Лечение SRD5A3-CDG симптоматическое.
2. Эластолизис 2-го типа, наследуемый по аутосомнорецессивному типу (ARCL-2A или ATP6V0A2-CDG, ATP6V1A-CDG и ATP6V1E1-CDG):
- Клинические проявления. ATP6V02-CDG представляет собой синдром множественных пороков развития, первоначально описанный как синдром дряблой кожи, а недавно выявленный как сочетание нарушений N- и О-гликозилирования. У пациентов при рождении наблюдается генерализованная неэластичная обвисшая кожа, гипотония, косоглазие, миопия, характерные черты лица и гиперподвижность суставов. Характерными чертами лица являются гипертелоризм, короткий нос, длинный подносовый желобок, скошенные книзу глазные щели с опущенными веками и отвисшие щеки. Поражение сердечно-сосудистой системы встречается редко, поражение ЦНС варьирует. Часто наблюдаются судороги и нарушения двигательного и речевого развития, также описаны случаи пациентов с нормальным интеллектом. Иногда присутствует нейросенсорная тугоухость.
У некоторых пациентов наблюдается гипоплазия червя мозжечка, а у нескольких детей на МРТ ГМ была описана булыжникоподобная дисгенезия и частичная пахигирия. Часто встречаются деформации скелета и низкий рост, а также поздно закрывающиеся роднички и/или брахидактилия и сколиоз. Нередко наблюдается дисплазия эмали. Характеристики кожи с возрастом самопроизвольно улучшаются. Фенотипы ATP6V1A-CDG и ATP6V1E1-CDG часто сопровождаются сердечно-сосудистыми симптомами и гиперхолестеринемией.
- Патофизиология. ATP6V0A2 представляет собой мембранную субъединицу протонной помпы комплекса везикулярной аденозинтрифосфатазы (В-АТФазы). Нарушение функции В-АТФазы приводит к изменению градиента pH в секреторном пути, а также воздействует на созревание и транспорт нескольких гликозилтрансфераз и эластических волокон (напр., эластина). ATP6V1A и ATP6V1E1 также относятся к субъединицам комплекса В-АТФазы. Они влияют на функцию ATP6V0A2 и приводят ко вторичному дефициту АТФазы. На это влияет как N-, так и О-гликозилирование. У некоторых пациентов наблюдаются легкие нарушения коагуляции и высокое содержание трансаминаз в сыворотке.
- Диагностика. Первичным скринингом для пациентов с подозрением на ATP6V0A2-CDG является анализ гликозилирования трансферрина с помощью изоэлектрической фокусировки (TIEF) или методом масс-спектрометрии. У большинства пациентов наблюдается паттерн 2-го типа (см. рис. 4), но также были описаны л/о-результаты у пациентов в возрасте до 6 нед. Аполипопротеин III-С (апоС-III) представляет собой секреторный гликопротеин муцинового типа, который является только О-гликозилированным. АпоС-III ИЭФ показывает паттерн гипергликозилирования у пациентов, даже если TIEF является л/о. При проведении биопсии кожи пациентов наблюдаются классические гист. изменения эластолиза с уменьшенными, короткими, аномальными и нечеткими эластическими волокнами.
ATP6V0A2-CDG наследуется АуР. Генетическое тестирование обычно выполняется путем прямого секвенирования. Точная заболеваемость неизвестна. Дефекты ATP6V1A и ATP6V1E1 были описаны сравнительно недавно.
- Лечение. При АуР-эластолизе 2-го типа лечение является симптоматическим. Наблюдается постоянное и самопроизвольное улучшение кожных симптомов, особенно при ATP6V0A2-CDG.
- Клинические проявления. Дефект Golgi-α1,2-маннозидазы (MAN1B1) первоначально описывался как синдром умственной отсталости в сочетании с дисморфическими признаками. У других пациентов были выявлены психомоторные нарушения, мышечная гипотония и втянутые соски в сочетании с ожирением туловища. Степень умственной отсталости весьма различна. Часто встречаются аутичное поведение, пищевые расстройства и агрессивное поведение. Зарегистрировано >30 пациентов.
- Патофизиология. Ген MAN1B1 кодирует маннозидазу аппарата Гольджи. Этот фермент играет ключевую роль в заключительной «обрезке» единиц маннозы в процессе синтеза гликанов в аппарате Гольджи. Гиперманнозилирование приводит к образованию патологических усеченных гликанов и развитию CDG II. Нарушение гликозилирования в сыворотке относительно умеренное. Повышенное содержание трансаминаз в сыворотке и нарушения коагуляции встречаются редко.
- Диагностика. У большинства пациентов наблюдается паттерн 2-го типа, характеризующийся незначительными отклонениями от нормы по результатам TIEF, но было описано несколько л/о случаев. При проведении MAN1B1-CDG в сыворотке обнаруживают характерные гибридные гликаны. В подозрительных случаях рекомендуют проводить прямое секвенирование, даже при нормальных результатах TIEF.
MAN1B1-CDG наследуется АуР. Точная частота встречаемости неизвестна; известно несколько взрослых пациентов.
- Лечение. Применяется только симптоматическая терапия.
4. Дефицит фосфоглюкомутазы-1 (PGM1-CDG):
- Клинические проявления. PGM1-CDG — расстройство, проявляющееся пороками развития срединной линии (расщелиной твердого нёба, синдромом Пьера Робена, раздвоение небного язычка), нарушением функции печени, гипогликемией и низким ростом почти у всех пациентов. Гипогликемия, как правило, связана с гиперинсулинизмом, возникающим на первом году жизни. С возрастом гиперинсулинизм может разрешаться. Кроме того, наблюдают кетотическую гипогликемию. У некоторых пациентов описаны холестаз, фиброз печени и даже цирроз. Кроме того, примерно у трети пациентов наблюдают проксимальную мышечную слабость и ДКМП. Последняя стала причиной летального исхода по меньшей мере в семи зарегистрированных случаях. Описаны также др. пороки развития, в т.ч. пороки развития сердца и скелета. Нередко нарушено заживление ран. Имеется очень высокий риск кровотечений во время хирургических вмешательств. Интеллект сохранен.
- Патофизиология. PGM1 — фермент, который играет очень важную роль в процессах гликогенолиза и гликолиза. Кроме того, данный фермент обеспечивает образование субстрата для сахаров, входящих в состав нуклеотидов, которые необходимы для нормального гликозилирования. PGM1 регулирует реакцию превращения глюкозо-1-фосфата и глюкозо-6-фосфата, проходящую в обоих направлениях. В период голодания это приводит к развитию гликогенозоподобного фенотипа (называемого также гликогенозом типа XIV, MIM 614921). PGM1-CDG воздействует на процессы гликозилирования, проходящие как в эндоплазматическом ретикулуме, так и в аппарате Гольджи, и приводит к развитию смешанного паттерна гликозилирования, объединяющего 1-й и 2-й тип.
Лабораторные отклонения от нормы характеризуются снижениями: факторов коагуляции и антикоагуляции, белка, связывающего инсулиноподобный фактор роста-3 (IGFBP3; англ. Insulin-like growth factor-binding protein 3), тироксинсвязывающего глобулина и ТТГ. Кроме того, отмечают отклонение от нормы уровней трансаминаз, гипогликемию и повышенные уровни КФК.
- Диагностика. Основным методом скрининга пациента с подозрением на PGM1-CDG является анализ гликозилирования трансферрина с помощью TIEF или методом масс-спектрометрии. У пациентов наблюдают смешанный паттерн, характеризующийся признаками 1-го и 2-го типа.
PGM1-CDG наследуется АуР. Это заболевание является одним из относительно распространенных CDG. Описано >40 пациентов. Можно исследовать уровни фермента в крови, но результаты исследования в фибробластах более достоверны. Доступен метод прямого секвенирования.
- Лечение. PGM1-CDG, по-видимому, является вторым по излечимости CDG после MPI-CDG. Предполагают, что D-галактоза восстанавливает баланс в отношении доступности разл. сахаров, входящих в состав нуклеотидов. Добавление в пищу D-галактозы в дозе 1 г/кг в сутки в течение нескольких недель значительно повышает активность гликозилирования несмотря на то, что паттерны TIEF до конца не нормализуются. На фоне такой терапии улучшаются показатели печеночных трансаминаз и антитромбина, а у некоторых пациентов и гормональный статус. Каким образом D-галактоза влияет на эпизоды гипогликемии и миопатию, до конца не ясно. На данный момент продолжаются крупномасштабные долгосрочные исследования по изучению диетотерапии.
5. Заболевания, сопровождающиеся нарушением гомеостаза аппарата Гольджи: ТМЕМ199-, CCDC115-, ATP6AP2-CDG и ATP6AP1-CDG:
- Клинические проявления. Указанные четыре нарушения невозможно различить по клиническим признакам и биохимическим показателям. При них описаны нарушения функции печени, сопровождающиеся холестазом, фиброзом и циррозом с печеночной недостаточностью. Нескольким пациентам потребовалась трансплантация печени. Фенотип напоминает болезнь Вильсона, в связи с низкими уровнями церулоплазмина и меди в сыворотке крови, однако кольца Кайзера-Флейшера отсутствуют. При CCDC115-CDG также часто наблюдаются отклонения со стороны НС. Исходы в отношении уровня интеллекта различны. Кроме того, могут наблюдаться такие отклонения, как гиперхолестеринемия и повышенные уровни ЩФ. При АТР6АР1-CDG также поражается иммунная система.
- Патофизиология. ТМЕМ199-, CCDC115-, ATP6AP1-CDG и ATP6AP2-CDG имеют большое значение для поддержания гомеостаза в аппарате Гольджи. Точные механизмы патогенеза пока не известны, однако предполагают, что вторичная дисфункция аппарата Гольджи влияет на нормальный процесс гликозилирования и приводит к его задержке.
- Диагностика. Первичным скринингом этих пациентов является анализ гликозилирования трансферрина с помощью TIEF или методом масс-спектрометрии. У пациентов наблюдают паттерн, характерный для 2-го типа (см. рис. 4). Результаты ИЭФ апоС-Ш отклоняются от нормы. При проведении MAN1B1-CDG в сыворотке обнаруживают характерные гибридные гликаны, но они не позволяют различить три дефекта. Для установления окончательного диагноза требуется мутационный анализ.
Для TMEM199-CDG и CCDC115-CDG характерен АуР тип наследования, а для ATP6AP1-CDG и АТР6АР2-CDG — Х-сцепленный.
- Лечение. Применяют симптоматическую терапию. Два пациента успешно перенесли трансплантацию печени.
6. Дефект транспортера марганца: SLC39A8-CDG:
- Клинические проявления. Это интересное нарушение первоначально описывалось как неврологическое заболевание с гипотонией, судорогами (гипсаритмией) и умственной отсталостью. У некоторых из описанных позже пациентов наблюдалась тяжелая дисплазия скелета с ризомелической хондродисплазией, краниосиностозом и карликовостью. Также может наблюдаться митохондриальная дисфункция (болезнь Лея, церебральная молочнокислая ацидемия, дистония).
- Патофизиология. SLC39A8 является мембранным транспортером, отвечающим за трансмембранный транспорт марганца (Мп). Дефицит SLC39A8 влияет на все Мп-зависимые ферменты и, следовательно, на разные части метаболизма. Поскольку некоторые гликозилтрансферазы (напр., β-1,4-галактозилтрансфераза) являются Mn-зависимыми, возникает вторичное гликозилирование Гольджи с дефектом гликозилирования 2-го типа. Низкий уровень марганца в сыворотке позволяет заподозрить диагноз, но этот признак не всегда присутствует у пациентов.
- Диагностика. Основным методом скрининга пациента с подозрением на SLC39A8-CDG является анализ гликозилирования трансферрина с помощью TIEF или методом масс-спектрометрии. У пациентов наблюдают паттерн, характерный для 2-го типа (см. рис. 4). Результаты MAN1B1-CDG позволяют предположить диагноз, но не позволяют отличить один дефект от другого. Для установления окончательного диагноза требуется анализ мутации.
- Лечение. Проводится симптоматическая терапия. Кроме того, у нескольких пациентов наблюдали улучшение биохимических и клинических показателей (улучшился контроль судорог) на фоне применения D-галактозы внутрь (1-3 г/кг в сутки).
д) Врожденные нарушения дегликозилирования:
1. Дефицит N-гликаназы-1 (дефект NGLY1):
- Клинические проявления. У пациентов с дефицитом NGLY1 нарушено гликозилирование, однако это не связано с дефицитом синтеза. Причиной скорее является дефицит расщепления гликопротеинов. Клинические проявления включают тяжелое поражение ЦНС, микроцефалию, умственную отсталость, судороги, невропатию, двигательные нарушения и мышечную гипотонию. Наличие алакримии, гиполакримии или халязиона с высокой степенью вероятности указывает на диагноз, однако проблемы со слезоотделением отмечаются не у всех пациентов. Кроме того, у пациентов отсутствуют прибавки МТ, ЗВУР и поражение печени. У некоторых пациентов наблюдают характерное овальное лицо с коротким носом, уплощенным профилем и гипертелоризм. М.б. также маскообразное лицо, имитирующее фенотип митохондриальных нарушений, особенно в сочетании с повышенными уровнями молочной кислоты сыворотки.
- Патофизиология. N-гликаназа отвечает за дегликирование неправильно свернутых N-гликопротеинов. Фермент необходим для расщепления гликанов, предшествующего расщеплению белков в эндоплазматическом ретикулуме. На фоне большого количества неправильно свернутых N-гликопротеинов усиливается стресс эндоплазматического ретикулума. Предполагают, что это является причиной повышения уровня молочной кислоты у некоторых пациентов. Кроме того, нередко повышаются уровни трансаминазы и АФП сыворотки.
- Диагностика. Результаты анализа изоформ трансферрина указывают на нормальный паттерн. Для установления окончательного диагноза требуется мутационный анализ. NGLY1-CDG является АуР-заболеванием. Большинство распространенных мутаций приходятся на с.1201А>Т/р. R401X. Точные показатели заболеваемости неизвестны, однако за несколько лет, прошедших с момента открытия заболевания, зарегистрировано >20 пациентов.
- Лечение. Для лечения пациентов с дефицитом NGLY1 используется только симптоматическая терапия.
е) Терапевтическое резюме. В большинстве случаев при CDG применяют только симптоматическую терапию. Доказано, что первоначально открытый метод лечения MPI-CDG с помощью перорального применения маннозы (1 г/кг в сутки) эффективен в отношении нарушений коагуляции и белковых энтеропатий, однако он не может предотвратить фиброз печени ни у одного из пациентов. У некоторых пациентов с MPI-CDG оказалась эффективной трансплантация печени. На фоне применения D-галактозы внутрь (1 г/кг в сутки) у пациентов с PGM1-CDG отмечают улучшение показателей трансаминаз сыворотки и коагуляции, а также положительный эффект в отношении эндокринной функции, однако полное восстановление гликозилирования невозможно. У пациентов с SLC39A8-CDG на фоне приема D-галактозы внутрь (1 г/кг в сутки) и препаратов марганца отмечают снижение частоты судорожных припадков.
Высокие дозы ингибиторов холинэстеразы успешно применяют для лечения врожденного миастенического синдрома при DPAGT1-CDG, GFPT1-CDG и GMPPB-CDG. Некоторые формы CDG поддаются контролю при трансплантации. К таким формам относятся DOLK-CDG (DK1-CDG, трансплантация сердца) PGM3-CDG (трансплантация гематопоэтических клеток), CCDC155-CDG (трансплантация печени).
При нарушениях, не описанных в данной главе, доступны и др. варианты лечения CDG. У пациентов с CAD-CDG отмечают значительное клиническое улучшение на фоне перорального применения уридина, особенно в отношении контроля судорожных припадков. У двух детей с дефектом SLC35C1-CDG наблюдали улучшение иммунных функций на фоне перорального применения фукозы. У пациентов с GNE-CDG отмечали значительное повышение мышечной силы на фоне терапии N-ацетилманнозоамином. В настоящее время продолжается несколько исследований по изучению диетотерапии при разл. формах CDG.