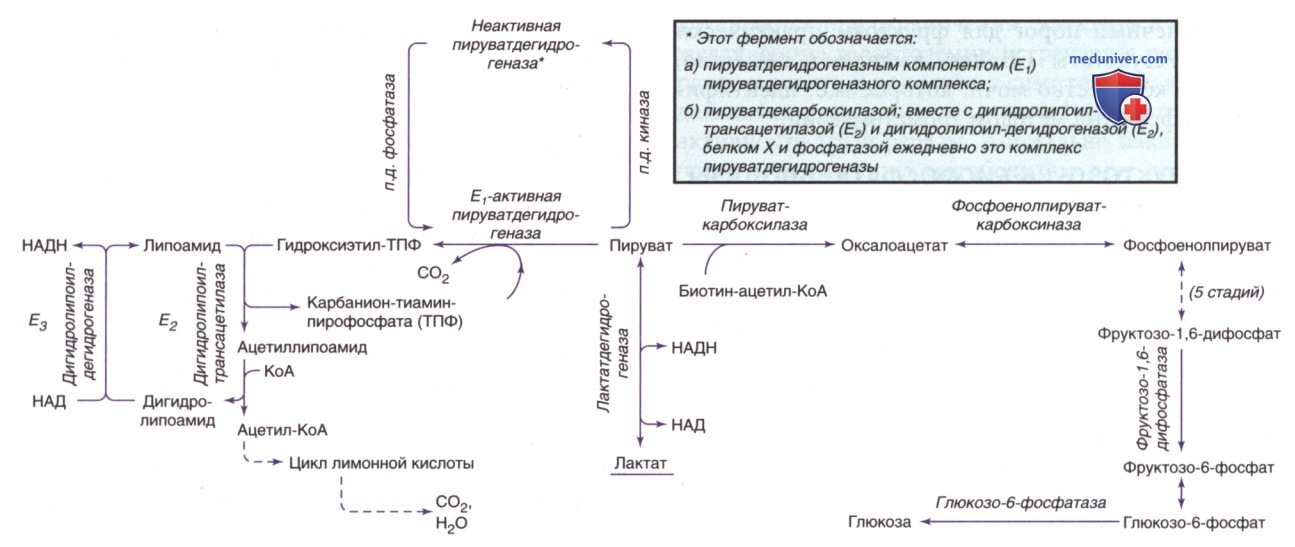
Лактатацидоз (тип В3) возникает при нарушениях углеводного обмена, которые препятствуют превращению пирувата в глюкозу через путь глюконеогенеза или в углекислый газ и воду через митохондриальные ферменты цикла Кребса. На рис. 1 изображены соответствующие пути обмена. Гликогеноз (GSD; англ. glycogen storage diseases) типа I, дефицит фруктозо-1,6-дифосфатазы и дефицит фосфоенолпируваткарбоксилазы представляют собой нарушения глюконеогенеза, связанные с лактатацидозом.
Рисунок 1. Ферментативные реакции углеводного обмена, дефицит которых может вызвать лактатацидоз, повышение пирувата или гипогликемию. Комплекс пируватдегидрогеназы включает, помимо E1, Е2 и Е3, дополнительный липоатсодержащий белок (не показан), называемый протеином X, и пируватдегидрогеназофосфатазу
Недостаточность пируватдегидрогеназного комплекса, дефекты дыхательной цепи и недостаточность пируваткарбоксилазы представляют собой нарушения в метаболизме пирувата, вызывающие лактатацидоз. Лактатацидоз (тип В3) также может возникать при дефектах окисления жирных кислот, органических ацидуриях или заболеваниях, нарушающих утилизацию биотина (тип В3) (табл. 2). Эти нарушения легко отличить по наличию аномальных профилей ацилкарнитина, аминокислот в крови и необычных органических кислот в моче.
Профили лактата, пирувата и ацилкарнитина в крови, а также наличие этих необычных органических кислот в моче должны определяться у младенцев и детей с необъяснимым ацидозом, особенно при увеличении анионной разницы.
Лактатацидоз, не связанный с недостаточностью ферментов, возникает при гипоксемии (лактатацидоз типа А). В этом случае, а также при дефектах дыхательной цепи концентрация пирувата в сыворотке крови может оставаться нормальной (<1,0 мг/дл, с повышенным соотношением лактат:пируват), тогда как пируват обычно повышается, когда лактатацидоз является результатом недостаточности ферментов глюконеогенеза или комплекса пируватдегидрогеназы (и лактат, и пируват повышены, а соотношение нормальное).
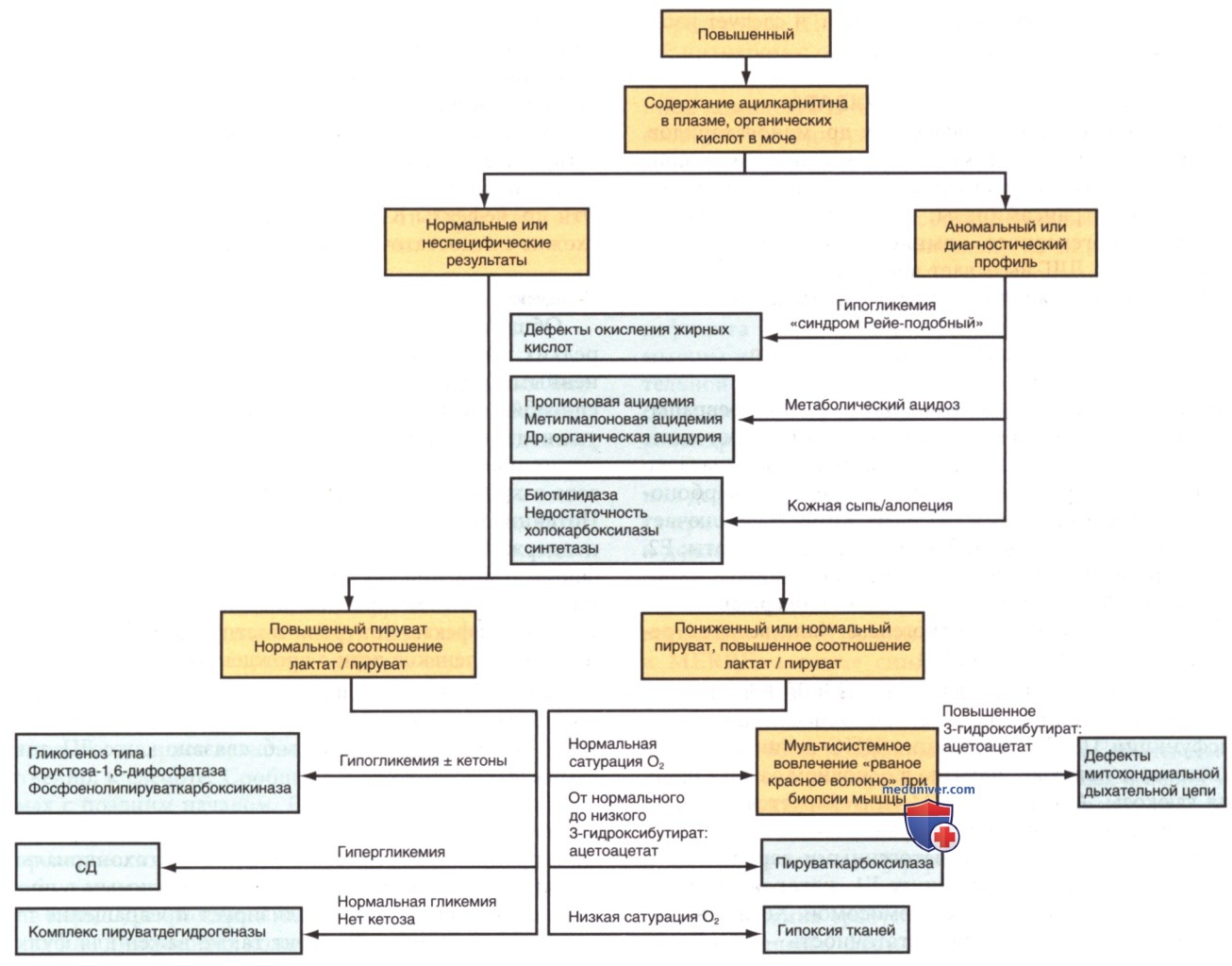
Лактат и пируват следует измерять в одном и том же образце крови и в нескольких образцах крови, полученных при наличии у пациента симптомов, поскольку лактатацидоз может возникать периодически. Рис. 2 представляет собой алгоритм дифференциальной диагностики лактатацидоза. Причинами лактатацидоза могут быть разл. основные заболевания (тип В1), ЛП или токсины (тип В2) (см. табл. 2).
1. Недостаточность глюкозо-6-фосфатазы (гликогеноз типа I). Гликогеноз типа I — единственный гликогеноз, сопровождающийся значительным лактатацидозом. Хронический метаболический ацидоз увеличивает риск развития у пациента остеопении, а острая гипогликемия с ацидозом при голодании может приводить к опасным для жизни состояниям.
2. Недостаточность фруктозо-1,6-дифосфатазы. Недостаточность фруктозо-1,6-дифосфатазы нарушает образование глюкозы из всех глюконеогенных предшественников, включая диетическую фруктозу. Гипогликемия возникает, когда запасы гликогена ограничены или истощены. Клинические проявления характеризуются опасными для жизни эпизодами ацидоза, гипогликемии, гипервентиляции, судорог и комы. Примерно в половине случаев недостаточность проявляется на 1-й неделе жизни.
У младенцев и маленьких детей приступы спровоцированы разл. инфекциями и гастроэнтеритом в случае уменьшения питания. Частота приступов с возрастом уменьшается. Лабораторные данные включают низкий уровень глюкозы в крови, высокий уровень лактата и мочевой кислоты и метаболический ацидоз. В отличие от ННФ, отвращения к сладкому обычно нет; функция почек и печени в норме.
Диагноз устанавливают, определяя недостаточность фермента при биопсии печени или кишечника. В некоторых случаях дефект фермента может также выявляться в лейкоцитах. Ген, кодирующий фруктозо-1,6-дифосфатазу (FBP1), расположен на хромосоме 9q22; патогенные варианты известны, что делает возможным выявление носителей и пренатальную диагностику. Лечение острых приступов заключается в коррекции гипогликемии и ацидоза путем в/в введения глюкозы; реакция обычно быстрая.
Избегание голодания, лечение инфекций, а также ограничение фруктозы и сахарозы в рационе могут предотвратить дальнейшие эпизоды заболевания. Для долгосрочной профилактики гипогликемии полезен медленно высвобождаемый углевод, напр. кукурузный крахмал. Выжившие пациенты в дальнейшем развиваются нормально.
3. Недостаточность фосфоенолпируваткарбоксикиназы. Фосфоенолпируваткарбоксикиназа (ФЕПКК) является ключевым ферментом в глюконеогенезе. Он катализирует превращение оксалоацетата в фосфоенолпируват (см. рис. 1). Недостаточность ФЕПКК является как недостаточностью митохондриального фермента, так и цитозольного фермента, кодируемых двумя разными генами.
О недостаточности ФЕПКК сообщалось лишь в нескольких случаях. Клинические признаки неоднородны, основными проявлениями являются гипогликемия, повышение лактата в крови, гепатомегалия, гипотония, нарушение и задержка развития. Может наблюдаться мультисистемное поражение с нервно-мышечным дефицитом, гепатоцеллюлярным повреждением, нарушением функции почек и кардиомиопатией. Диагноз основывается на снижении активности ФЕПКК в печени, фибробластах или лимфоцитах. Фибробласты и лимфоциты не подходят для диагностики цитозольной формы дефицита ФЕПКК, потому что эти ткани обладают только митохондриальной ФЕПКК.
Чтобы избежать гипогликемии, пациенты должны получать лечение углеводами с медленным высвобождением, такими как кукурузный крахмал, и следует избегать голодания.
б) Нарушения метаболизма пирувата. Пируват образуется из глюкозы и др. моносахаридов, из лактата и аланина. Он метаболизируется с помощью четырех основных ферментных систем: лактатдегидрогеназы, аланинтрансаминазы, пируваткарбоксилазы и пируватдегидрогеназного комплекса. Недостаточность М-субъединицы ЛДГ вызывает непереносимость физических упражнений и миоглобинурию.
1. Недостаточность пируватдегидрогеназного комплекса. После попадания в митохондрии пируват превращается в ацетил-КоА комплексом пируватдегидрогеназы (КПДГ), который катализирует окисление пирувата до ацетил-КоА, который затем входит в цикл трикарбоновой кислоты для производства АТФ. Комплекс включает пять компонентов: Е1, декарбоксилаза а-кетокислоты; Е2, дигидролипоилтрансацилаза; ЕЗ, дигидролипоилдегидрогеназа; белок X, дополнительный липоатсодержащий белок; и фосфатаза пируватдегидрогеназа. Чаще всего встречается дефект Е1 (см. рис. 1).
Недостаточность КПДГ является наиболее частым из расстройств, приводящих к молочнокислой ацидемии и дисфункции ЦНС. Дисфункция ЦНС возникает из-за того, что мозг получает энергию в основном за счет окисления глюкозы. Ацетил-КоА ГМ синтезируется по большей части исключительно из пирувата.
Дефекты Е1 вызываются патогенными вариантами в гене, кодирующем α-субъединицу Е1, которая является сцепленной с доминантной Х-хромосомой. Хотя ген сцеплен с Х-хромосомой, его недостаточность — проблема как для мужчин, так и для женщин, несмотря на только один аллель Е1а у женщин, переносящих вариант.
- Клинические проявления. Недостаточность КПДГ имеет широкий спектр проявлений, от наиболее тяжелых неонатальных до легкой формы с поздним началом. Неонатальное начало связано со смертельным лактатацидозом, кистозными поражениями белого вещества, агенезией мозолистого тела и наиболее серьезной недостаточностью ферментов. Младенческая форма м.б. летальной или связанной с задержкой психомоторного развития и хроническим лактатацидозом, кистозными поражениями ствола мозга и базальных ганглиев, а также патологическими особенностями, напоминающими болезнь Ли.
Неврологические симптомы при КПДГ можно разделить на две группы: аномалии развития мозга, наблюдаемые как у мужчин, так и у женщин, и поражение мозга и эпилепсия, наблюдаемые только у пациентов мужского пола. У детей старшего возраста, обычно мальчиков, могут наблюдаться менее выраженный ацидоз, повышенная активность ферментов и атаксия при высокоуглеводной диете. Умственное развитие — нормальное. Пациенты любого возраста могут отличаться нарушением строения лица, сходным с признаками фетального алкогольного синдрома.
Дефекты Е2 и протеина Х-липоата встречаются редко и приводят к тяжелой задержке психомоторного развития. Дефект липоамиддегидрогеназы Е3 приводит к недостаточной активности не только в КПДГ, но и в комплексах α-кетоглутарата и разветвленноцепочечной дегидрогеназы кетокислоты. Этот недостаток чаще встречается у евреев-ашкенази. Активные формы кислорода, генерируемые патогенными вариантами, ответственными за недостаток липоамиддегидрогеназы, на самом деле могут объяснять определенные характеристики заболевания и предполагать полезность антиоксидантной терапии.
Сообщалось также о дефиците фосфатазы пируватдегидрогеназы. Эти др. дефекты КПДГ имеют клинические проявления, схожие с недостаточностью, вызванной Е1.
- Лечение. Общий прогноз неблагоприятный, за исключением редких пациентов, у которых варианты связаны с измененным сродством к пирофосфату тиамина и которые способны реагировать на лечение тиамином. Поскольку углеводы могут усугубить лактатацидоз, рекомендуется кетогенная диета. Диета призвана снижать уровень лактата в крови; долгосрочная польза для пациента неясна. Потенциальная стратегия лечения состоит в том, чтобы поддерживать любой остаточный КПДГ в его активной форме путем приема внутрь дихлррацетата, ингибитора киназы Е1.
У некоторых пациентов наблюдались положительные эффекты контроля постпрандиального лактатацидоза. Маленькие дети с врожденным ацидозом обычно хорошо переносят дихлорацетат, но его продолжающееся воздействие может приводить к периферической невропатии — состоянию, которое м.б. связано как с ЛП, так и с самим заболеванием.
2. Недостаточность пируваткарбоксилазы. Пируваткарбоксилаза является митохондриальным биотин-содержащим ферментом, необходимым в процессе глюконеогенеза; он катализирует превращение пирувата в оксалоацетат. Фермент также важен для функции цикла Кребса в качестве поставщика оксалоацетата и участвует в липогенезе и образовании заменимых аминокислот. Клинические проявления этой недостаточности варьировались от тяжелого лактатацидоза у новорожденных, сопровождающегося гипераммониемией, цитруллинемией и гиперлизинемией (тип В), до позднего легкого и умеренного лактат-ацидоза и задержки развития (тип А). В обоих типах у выживших пациентов, как правило, наблюдалась тяжелая задержка психомоторного развития с судорогами, спазмами и микроцефалией.
У некоторых пациентов выявляются патологические изменения ствола мозга и базальных ганглиев, напоминающие болезнь Ли. Клиническая тяжесть коррелирует с уровнем остаточной активности фермента. Также была описана «доброкачественная» форма недостаточности пируваткарбоксилазы, характеризующаяся повторяющимися приступами лактатацидоза и легкими неврологическими нарушениями (тип С). Лабораторные данные характеризуются повышенным уровнем лактата, пирувата, аланина в крови и кетонурией. В случае типа В уровни аммиака, цитруллина и лизина в крови также повышены, что может указывать на первичный дефект цикла мочевины. Этот механизм, вероятно, вызван истощением оксалоацетата, что приводит к снижению уровня аспартата, субстрата аргининосукцинатсинтазы в цикле мочевины.
Ген пируваткарбоксилазы (PC) расположен на хромосоме 11q13.4-q13.5, и было идентифицировано ок. 15 патогенных вариантов.
Лечение заключается в отказе от голодания и употреблении углеводной пищи перед сном. Во время острых эпизодов лактатацидоза пациенты должны постоянно получать глюкозу в/в. Добавки аспартата и цитрата восстанавливают нарушения обмена веществ; неизвестно, может ли это лечение предотвратить неврологический дефицит. Предпринята попытка трансплантации печени; ее польза остается неизвестной. Диагноз недостаточности пируваткарбоксилазы ставится путем измерения активности ферментов в печени или культивируемых фибробластах кожи, и его необходимо дифференцировать от дефицита холокарбоксилазы синтазы или биотинидазы.
3. Вторичная недостаточность пируваткарбоксилазы по отношению к недостаточности холокарбоксилазосинтазы или биотинидазы. Недостаточность холокарбоксилазосинтазы или биотинидазы, которые являются ферментами метаболизма биотина, приводит к недостаточности множественных карбоксилаз (пируваткарбоксилазы и других биотин-требующих карбоксилаз и метаболических реакций), а также к клиническим проявлениям, которые соответствуют картине множественной недостаточности карбоксилаз и включают дерматит, лактатацидоз и алопецию.
Течение дефицита холокарбоксилазосинтазы или биотинидазы м.б. длительным, с периодическим обострением хронического лактатацидоза, задержкой развития, судорогами и гипотонией, ведущими к спастичности, летаргии, коме и смерти. Дисфункция слухового и зрительного нервов может привести к глухоте и, соответственно, к слепоте. Сообщалось также о более легких формах с поздним началом. Лабораторные исследования включают метаболический ацидоз и аномальное содержание органических кислот в моче. При недостаточности холокарбоксилазосинтазы концентрация биотина в плазме и моче в норме. Диагностика может проводиться на фибробластах или лимфоцитах кожи с помощью анализа активности холокарбоксилазосинтазы, а в случае биотинидазы в сыворотке крови — с помощью скрининга пятна крови.
Лечение состоит из добавок биотина в дозе 5-20 мг/сут; как правило, эффективно, если лечение начинается до развития повреждения ГМ. У пациентов, выявленных в результате скрининга новорожденных и получавших биотин, симптомы отсутствуют.
Недостаточность обоих ферментов является АуР-заболеванием. Распространенность недостаточности холокарбоксилазосинтазы составляет 1:87 000 живорождений. Ген ХКС и биотинидаза расположены на хромосомах 21q22 и Зр25 соответственно. Выявлены этнически специфические патогенные варианты в гене ХКС. Два распространенных патогенных варианта (del7/ins3 и p.R538C) в биотинидазе составляют 52% всех патогенных аллелей у пациентов с симптомами недостаточности биотинидазы.
4. Дефекты митохондриальной дыхательной цепи (болезнь окислительного фосфорилирования). Митохондриальная дыхательная цепь катализирует окисление энергетических уровней молекул и передает электроны молекулярному кислороду с сопутствующей трансдукцией энергии в аденозинтрифосфат (окислительное фосфорилирование). Дыхательная цепь вырабатывает АТФ из аденозиндифосфата и неорганического фосфата, используя энергию электронов, переносимых никотинамидадениндинуклеотидом (НАДН) или флавинадениндинуклеотидом, а также включает пять специфических комплексов (I: НАДН-коэнзим Q-редуктаза; II: сукцинат — кофермент Q-редуктаза; III: кофермент QH2-цитохром-с-редуктаза; IV: цитохром-с оксидаза; V: АТФ-синтаза).
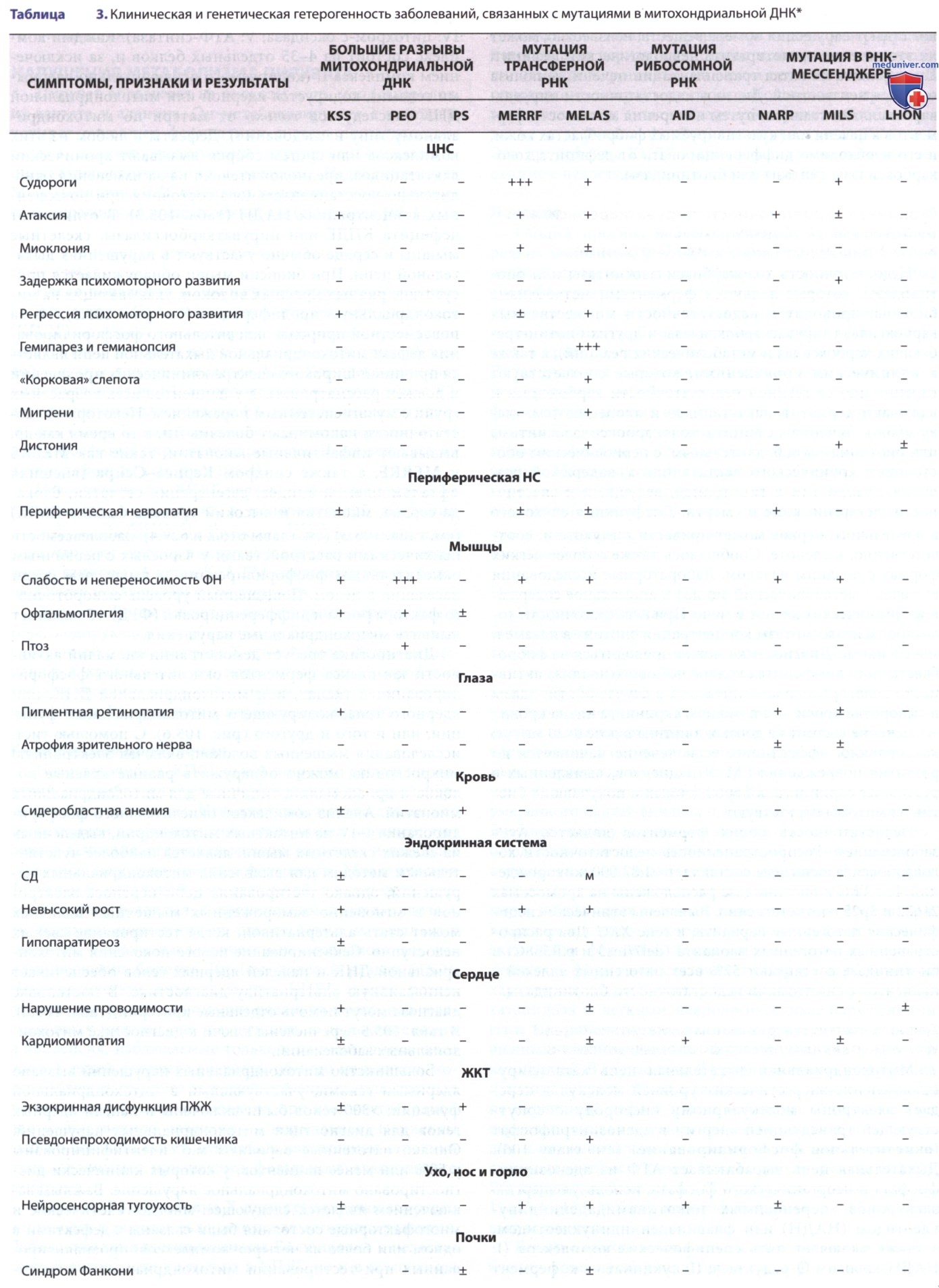
Каждый комплекс состоит из 4-35 отдельных белков и, за исключением комплекса II (кодируемого исключительно ядерными генами), кодируется ядерной или митохондриальной ДНК (наследуется только от матери по митохондриальному типу наследования). Дефекты в любом из этих комплексов или систем сборки вызывают хронический лактатацидоз, предположительно из-за изменения окислительно-восстановительного состояния при повышенных концентрациях НАДН (табл. 3). В отличие от дефицита КПДГ или пируваткарбоксилазы, скелетные мышцы и сердце обычно участвуют в нарушениях дыхательной цепи. При биопсии мышц обнаруживается присутствие рваных красных волокон, указывающих на митохондриальную пролиферацию (см. рис. 2).
Из-за повсеместной природы окислительного фосфорилирования дефект митохондриальной дыхательной цепи является причиной широкого спектра клинических проявлений и должен рассматриваться у пациентов всех возрастных групп с мультисистемным поражением. Некоторые недостаточности напоминают болезнь Ли, в то время как др. вызывают инфантильные миопатии, такие как MELAS и MERRF, а также синдром Кернса-Сейра (внешняя офтальмоплегия, ацидоз, дегенерация сетчатки, блокада сердца, миопатия и высокий уровень белка в СМЖ) (см. табл. 3). Заболеваемость психическими расстройствами у взрослых с первичным окислительным фосфорилированием выше, чем среди населения в целом.
Повышенный уровень сывороточного фактора роста и дифференцировки (ФРД)-15 помогает выявить митохондриальные нарушения.
Диагностика требует демонстрации аномалий активности комплекса ферментов окислительного фосфорилирования в тканях, или митохондриальной ДНК, или ядерного гена, кодирующего митохондриальные функции, или и того и другого (рис. 3). С помощью гист. исследования мышечных волокон, включая электронную микроскопию, можно обнаружить рваные красные волокна и др. аномалии, типичные для митохондриальных миопатий. Анализ комплексов окислительного фосфорилирования I-IV из интактных митохондрий, выделенных из свежих скелетных мышц, является наиболее чувствительным методом для выявления митохондриальных нарушений; однако тестирование цепи переноса электронов в мгновенно замороженных мышечных волокнах может стать альтернативой, когда тестирование свежих недоступно.
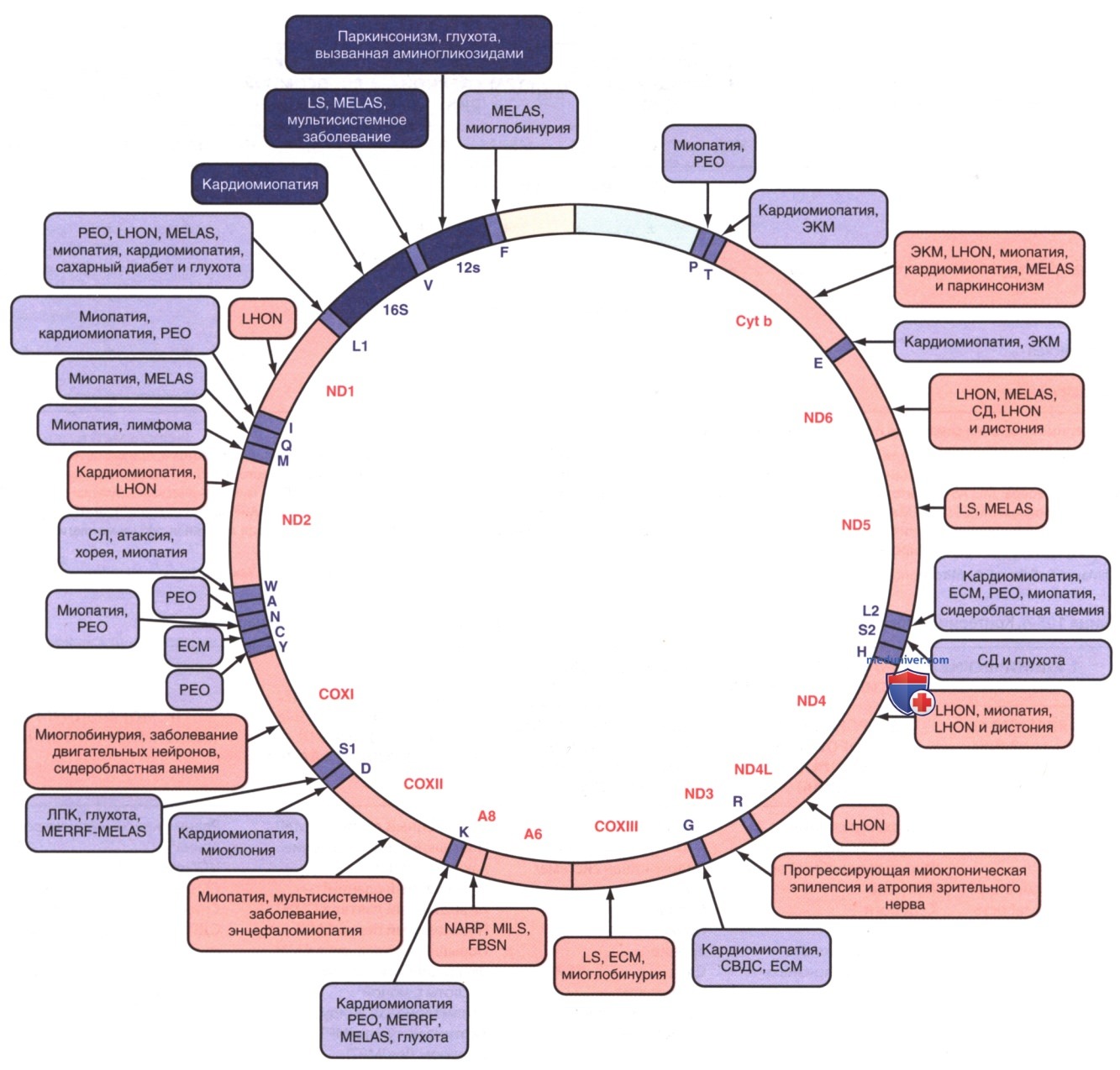
Рисунок 3. Мутации в митохондриальном геноме человека, которые вызывают заболевания. Нарушения, которые часто или явно связаны с мутациями в конкретном гене, выделены жирным шрифтом. Заболевания, вызванные мутациями, нарушающими синтез митохондриального белка, показаны синим цветом. Заболевания, вызванные мутациями в генах, кодирующих белок, показаны красным цветом. ЕСМ — энцефаломиопатия; FBSN — наследственный двусторонний стриарный некроз; LHON — наследственная оптическая невропатия Лебера; LS — синдром Лея; MELAS — митохондриальная энцефаломиопатия, лактоацидоз и инсульты; MERRF — миоклоническая эпилепсия с рваными красными волокнами; MILS — синдром Лея, наследуемый по материнской линии; NARP — невропатия, атаксия и пигментный ретинит; РЕО — прогрессивная внешняя офтальмоплегия; РРК — ладонно-подошвенная кератодермия; СВДС — синдром внезапной детской смерти
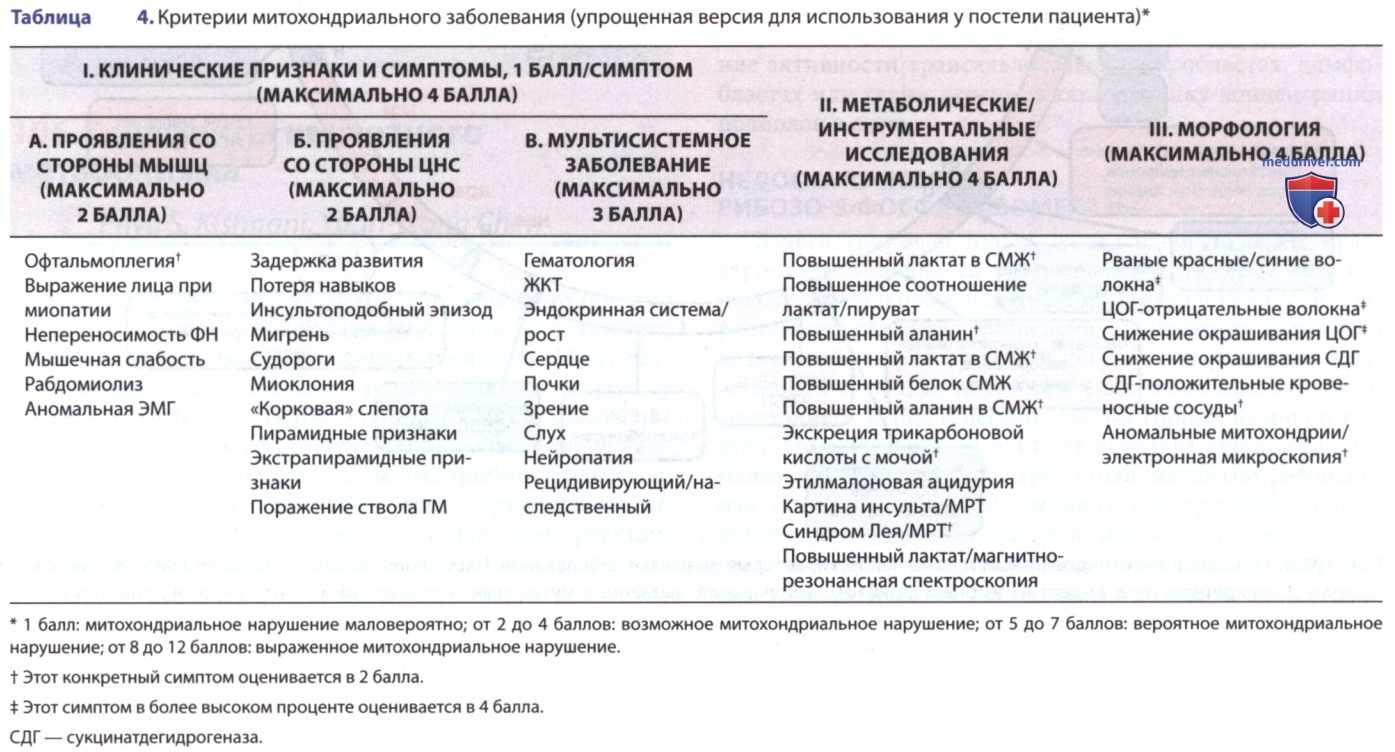
Секвенирование нового поколения митохондриальной ДНК и панелей ядерных генов обеспечивает неинвазивную альтернативу диагностике. В постановке диагноза могут помочь отдельные критерии (табл. 4). В табл. 5 перечислены ключи к диагностике митохондриальных заболеваний.
Большинство митохондриальных нарушений вызвано ядерными генами, участвующими в митохондриальной функции; >300 генов были включены в панели ядерных генов для диагностики митохондриальных нарушений. Однако патогенные варианты м.б. идентифицированы у 50% или менее пациентов, у которых клинически диагностировано митохондриальное нарушение. Важным заключением является следующее: многие генетические и многофакторные состояния были связаны с дефектами в одном или более из четырех комплексов, проанализированных при тестировании митохондриального окислительного фосфорилирования. Последние состояния характеризуются так называемой вторичной дисфункцией митохондрий, поскольку сами по себе они не считаются митохондриальными нарушениями.
Лечение остается в основном симптоматическим и существенно не меняет исход заболевания. Некоторые пациенты реагируют на добавки с кофакторами, обычно на кофермент Q10 ± L-карнитин в фармакологических дозах. Добавление креатина моногидрата и α-липоевой кислоты может принести значительную пользу. EPI-743 представляет собой парабензохиноноподобный агент, обладающий защитной активностью против окислительного повреждения; это довольно многообещающий агент при лечении митохондриальных нарушений, включая синдром Ли.
5. Болезнь Ли (подострая некротическая энцефаломиелопатия). Болезнь Ли — гетерогенное неврологическое заболевание, характеризующееся демиелинизацией, глиозом, некрозом, относительной сохранностью нейронов и пролиферацией капилляров в определенных областях ГМ. У пациентов с болезнью Ли часто наблюдаются проблемы с кормлением и глотанием, отмечается задержка роста и развития. Проявления сильно варьируются и могут включать судороги, измененное сознание, выпот в перикарде и дилатационную кардиомиопатию. Диагноз обычно подтверждается рентгенологическими или патологическими данными о симметричных поражениях базальных ганглиев, ствола мозга и субталамических ядер. У пациентов с болезнью Ли имеются дефекты нескольких ферментных комплексов.
Дисфункция цитохром-с-оксидазы (комплекс IV) является наиболее часто встречающимся дефектом, за ней следуют НАДН-кофермент Q-редуктаза (комплекс I), КПДГ и пируваткарбоксилаза. Патогенные варианты в ядерном гене SURF1, который кодирует фактор, участвующий в биогенезе цитохром-с-оксидазы, и варианты митохондриальной ДНК в кодирующей области аденозинтрифосфатазы-6, были зарегистрированы у пациентов с болезнью Ли из-за недостаточности комплекса IV. Наиболее распространенным вариантом митохондриальной ДНК при болезни Ли является вариант T8993G в МТ-АТР6. Прогноз болезни Ли неутешительный. В исследовании 14 случаев было 7 летальных в возрасте до 1,5 года.
Сообщалось также о лактатацидозе, гипогликемии и энцефалопатии у пациентов с недостаточностью переносчика тиамина и с пиридоксин-зависимой эпилепсией. Оба расстройства должны разрешиться за счет приема тиамина и пиридоксина соответственно.