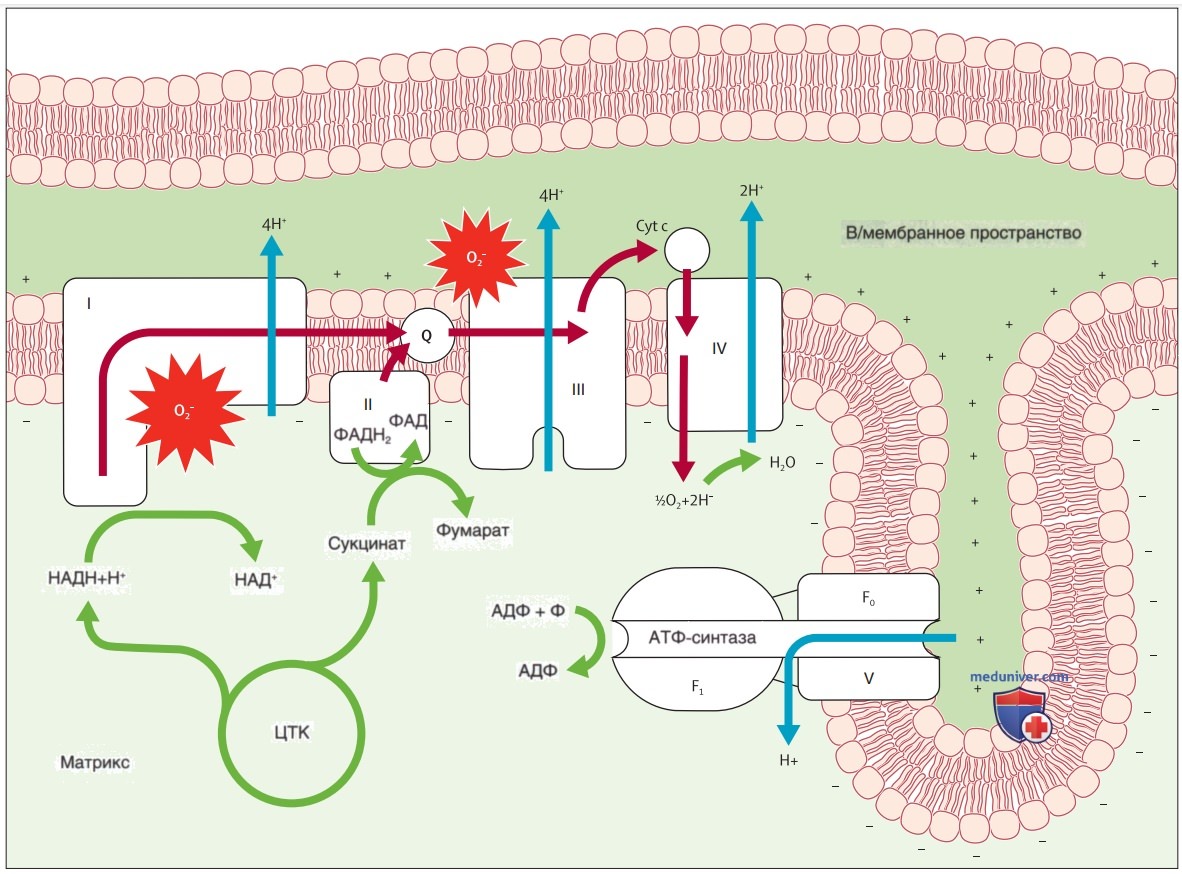
а) Обзор митохондриальных заболеваний. Митохондриальные заболевания — это мультисистемные состояния, характеризующиеся энергетическим дефицитом и обширной клинической и генетической неоднородностью. Их лучшее понимание заключается в признании того, что митохондрии функционируют как биологические «элементы питания» или «батареи», производя хим. энергию в форме АТФ за счет аэробного метаболизма поступивших питательных в-в производных восстановительных эквивалентов посредством интегрированной функции 5-комплексной митохондриальной дыхательной цепи (ДЦ) (рис. 1).
Рисунок 1. Цепь переноса электронов. Цепь переноса электронов состоит из четырех белковых комплексов (I-IV), связанных с 5-м (V), несвязанным комплексом — АТФ-синтазой. Вместе эти пять комплексов известны как дыхательные цепи (ДЦ) и являются местом, где происходит окислительное фосфорилирование (OXPHOS) для выработки энергии. Транспортная цепь принимает электроны от НАДН (комплекс I) или ФАДН2 (комплекс II), которые были произведены в результате гликолиза, образования ацетил-кофермента А и ЦТК (зеленые стрелки). Электроны перетекают от одного комплекса к другому (красные стрелки) за счет окислительно-восстановительного потенциала каждого комплекса и при движении по цепочке теряют небольшое количество энергии. Три из четырех комплексов действуют как насосы, приводимые в движение потоком электронов, перемещая ионы Н+ из матрицы во в/мембранное пространство (синие стрелки). Такое накачивание создает градиент концентрации и электрохим. силу, которая используется АТФ-синтазой для производства АТФ. В нормальных условиях этот механизм обеспечивает присутствие практически всего (90%) АТФ в клетке. Однако небольшая часть электронов покидает цепь переноса электронов даже при нормальных условиях и может реагировать с кислородом и комплексами I и III с образованием супероксида (О2-). АДФ — аденозиндифосфат; АТФ — аденозинтрифосфат; Cyt с — цитохром с; Q — коэнзим Q; НАДН — никотинамиддинуклеотид; Pi — неорганический фосфат; ЦТК — цикл трикарбоновых кислот; ФАДН2 — динуклеотид 1,5-дигидро-флавинаденин
Митохондрии также играют важную роль в таких процессах, как регулирование гомеостаза кальция, разл. аспектов промежуточного метаболизма питательных в-в, метаболизма нуклеотидов и окислительного стресса. Нарушение данных процессов приводит к формированию заболеваний. Первично митохондриальное заболевание является результатом недостаточной функции ДЦ, которая м.б. вызвана мутациями в генах, кодирующих субъединицы ДЦ, факторы сборки или кофакторы, компоненты метаболизма и поддержания митохондриальной ДНК (мтДНК) либо множество др. основных метаболических процессов, происходящих в митохондриях.
В митохондриальном протеоме разл. тканей существует 1500 белков с вариантами >350 уникальных генов как в ядерном, так и в митохондриальном геномах, которые уже считаются одной из причин развития митохондриальных заболеваний человека.
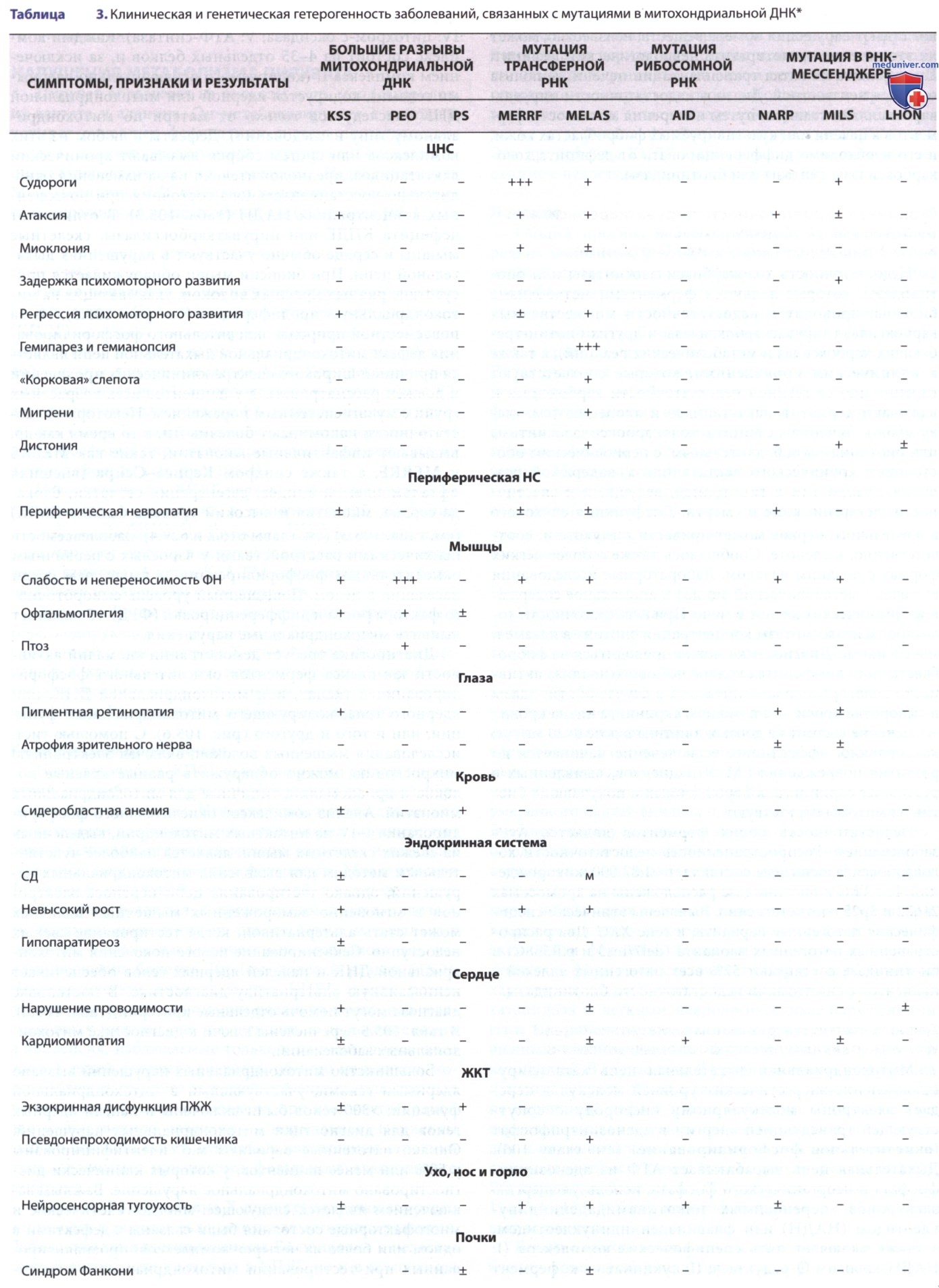
Первичные (наследственные) митохондриальные заболевания, совокупно признанные наиболее распространенной группой наследственных метаболических заболеваний, имеют минимальную суммарную распространенность 1:4300 человек в любом возрасте. Кроме того, вторичная дисфункция митохондрий широко вовлечена в патогенез множества сложных заболеваний, начиная от метаболического синдрома и заканчивая ишемически-реперфузионным повреждением после инсульта и нейродегенеративными заболеваниями. Поражение органов с высоким потреблением энергии при митохондриальных заболеваниях может клинически проявляться в виде тяжелых нарушений со стороны НС, патологий сердца, миопатий, нарушений со стороны почек, печени, эндокринной и иммунной систем, ЖКТ, снижения слуха и зрения, а так же общей дестабилизации метаболизма с лактоацидозом (рис. 2) (см. табл. 2, 3).
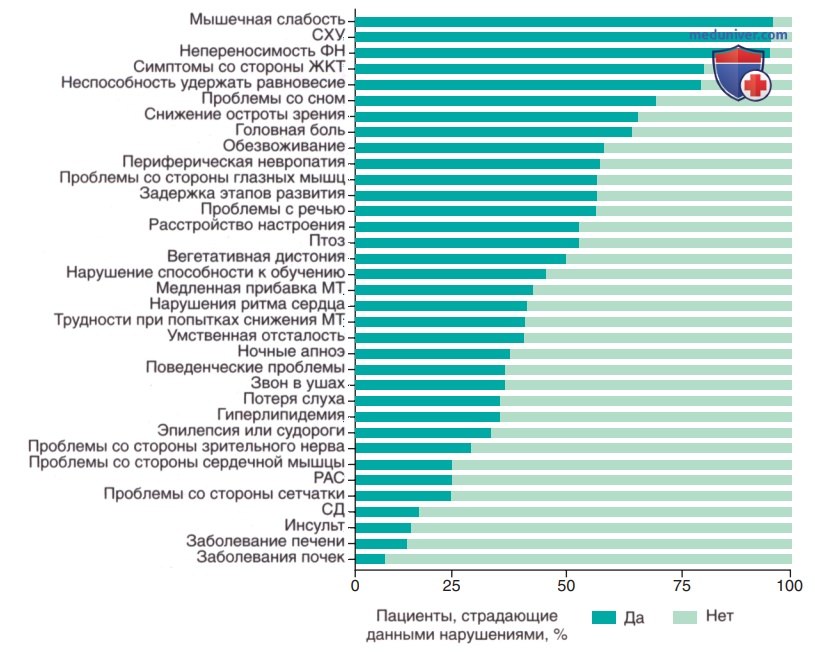
Рисунок 2. Симптомы у группы пациентов с митохондриальными заболеваниями. Согласно собственной оценке пациентов, участвовавших в программе RDCRN, пятью наиболее распространенными симптомами были мышечная слабость, синдром хронической усталости (СХУ), непереносимость физической нагрузки (ФН), неспособность удержать равновесие и проблемы со стороны желудочно-кишечного тракта (ЖКТ)
При большинстве митохондриальных нарушений клиника может варьироваться в зависимости от возраста пациента или конкретного гена или аллеля. К особо частым клиническим синдромам митохондриальных заболеваний у детей относятся синдром Лея (причиной развития которого являются нарушения в >90 генов), синдром истощения мтДНК (для которого существуют десятки причинных генов), синдромы делеции мтДНК (Пирсона, Кернса-Сейра), первичный лактоацидоз и недостаточность пируватдегидрогеназы. Общие клинические признаки у детей, присутствующие по меньшей мере у 90% пациентов, включают усталость, непереносимость физических упражнений, слабость, проблемы с ЖКТ, атаксию и задержку развития.
Т.о., митохондриальные заболевания могут встречаться в практике врачей всех мед. специальностей.
Пациенты, у которых предполагают митохондриальные заболевания, нередко проходят через «диагностическую одиссею» — как клиническую, так и генетическую. Их обширная фенотипическая гетерогенность при отсутствии общего биомаркера [тест на ростовой фактор дифференцировки-15 (GDF-15; англ. growth and differentiation factor 15)] м.б. одним из скрининговых тестов, результат которого бывает повышен при некоторых митохондриальных миопатиях, особенно связанных с делециями или истощением мтДНК наряду с лактоацидозом) представляет собой проблему для доступной и точной клинической диагностики митохондриальных расстройств во многих медучреждениях.
Точно так же их обширная генетическая гетерогенность, включая уже известные ядерные гены (>300) и 37 генов мтДНК и, предположительно, еще десятки или сотни генов, вызывающих болезнь генов ядерного генома, может затруднить точный генетический диагноз для отдельного пациента. Диагностическая неопределенность может дополнительно усугубляться плохой корреляцией «генотип-фенотип» и вариативностью клинических проявлений отдельных генных нарушений, высокой гетерогенностью локусов (т.е. множественными генами, нарушения в которых являются причиной разл. заболеваний) для сходных клинических фенотипов, неполной пенетрантностью для некоторых заболеваний генов, разл. факторами стресса в повседневной жизни или воздействием окружающей среды, которое может обострить конкретные заболевания у детей, а также уникальные биологические аспекты материнской наследственности для подмножества митохондриальных заболеваний, вызванных мутациями гена мтДНК.
б) Когда следует заподозрить митохондриальное заболевание? По причине отсутствия способности генерировать энергию в клетках митохондриальные заболевания могут поражать любую систему органов в любом возрасте (см. рис. 2). Митохондриальное заболевание следует подозревать при наличии классических симптомов или при появлении необъяснимых симптомов в трех или более органах, которые явно не связаны между собой. У пациентов может наблюдаться широкий спектр симптомов, включая усталость, мышечную слабость, непереносимость физических упражнений, метаболические инсульты, судороги, кардиомиопатию, аритмию, нарушения развития или когнитивные нарушения, РАС, СД, др. эндокринопатии (надпочечников, ЩЖ), дизавтономию и аутоиммунные нарушения, а также нарушения слуха, зрения, роста, патологии печени, ЖКТ или нарушения функции почек.
Несмотря на то что отдельные пациенты могут иметь один или несколько симптомов и изменение тяжести проявлений в течение времени, у большинства пациентов с первичным митохондриальным заболеванием клинические проявления со временем прогрессируют. В исследовании пациентов с митохондриальными заболеваниями количество клинически значимых симптомов на одного пациента колебалось в диапазоне от 7 до 35 и в среднем составляло 16. При рассмотрении диагноза необходимо понимать, что большинство симптомов митохондриального заболевания, скорее всего, связаны с функциональными проблемами, нежели со структурными.
При ДД митохондриальных заболеваний с др. сходными заболеваниями рекомендовано проведение нескольких скрининговых лабораторных исследований общих биохим. маркеров, при этом исследования необходимо проводить на начальном этапе диагностики, в периоде обострения заболевания или декомпенсации. Скрининговые исследования метаболизма на основе анализа крови включают комплексную хим. панель, полный клинический анализ крови, количественный анализ аминокислот в плазме, анализ на карнитин (общий, свободный, ацилкарнитиновый профиль), аммиак, КФК и исследования для выявления вторичных проявлений митохондриальных заболеваний (напр., скрининг ЩЖ, определение липопротеинового профиля, уровня HbA1c).
Скрининговые исследования метаболизма на основе исследований мочи включают ОАМ, количественный анализ органических кислот и количественный анализ аминокислот в моче. Следует также рассмотреть возможность проведения скрининга на врожденные нарушения процессов гликозилирования или дефицит витаминов, которые в некоторых случаях могут иметь клинические признаки, частично совпадающие с митохондриальными заболеваниями. Лактатацидоз не является высокочувствительным или специфичным признаком первичного митохондриального заболевания.
В то же время на первичное митохондриальное заболевание могут указывать повышение содержания лактата и пирувата в крови, соотношения лактат:пируват, аланина, отношения аланина к лизину (>3) и аланина к фенилаланину и тирозину в сумме (>4), а также анионный разрыв. Биохим. изменения, дополнительно указывающие на митохондриальное заболевание, могут включать вторичное нарушение окисления жирных кислот с повышением содержания дикарбоновых кислот в ацилкарнитиновом профиле, увеличение количества аминокислот с разветвленной цепью и пролина при анализе аминокислот в плазме, увеличение промежуточных продуктов цикла трикарбоновых кислот и экскрецию лактата с мочой (определяемую по анализу органических кислот), генерализованную аминоацидурию по аминокислотному анализу мочи. Полезным скрининговым тестом на определение миопатий, вызванных митохондриальным истощением, является GDF-15.
Точно так же при ДД митохондриальных заболеваний важно проводить полноценное клиническое обследование с целью выявления наиболее распространенных, высокопатологичных и меняющихся признаков митохондриального заболевания. Поскольку у многих людей с митохондриальными заболеваниями возникают проблемы со зрением (снижение остроты зрения, которое невозможно исправить с помощью очков, светобоязнь или никталопия с ухудшением периферического зрения, связанные с заболеванием сетчатки или атрофией зрительного нерва, офтальмоплегией, птозом), слухом (высокочастотная нейросенсорная потеря слуха), а также сердцем (аритмия, блокада проводимости, кардиомиопатия), рекомендуется тщательная оценка поражения этих высокоэнергетических систем.
Важна неврологическая оценка, потому что у многих пациентов с митохондриальными заболеваниями наблюдается ряд заболеваний центральной (метаболический инсульт в корковом или глубоком сером в-ве, включая базальные ганглии, средний мозг и/или ствол мозга, изменения белого в-ва, судороги, атаксия, двигательное расстройство, мигрень, когнитивные изменения), периферической (аксональная сенсомоторная невропатия) или дисфункция ВНС. Для подтверждения диагноза м.б. полезны МРТ, спектроскопия и в некоторых случаях ЭМГ или исследование скорости распространения возбуждения. Необходимо проведение функциональных проб для оценки возможностей пациента и последующего их консультирования с целью подбора индивидуальных и безопасных физических упражнений. Кроме того, изменения функциональных показателей (напр., снижение максимального потребления кислорода) могут указывать на митохондриальную дисфункцию.
Исследование сна может быть полезно для людей с дисфункцией сна, поскольку нарушения сна могут имитировать симптомы митохондриального заболевания. Проблемы со сном являются распространенными и потенциально поддаются лечению при митохондриальных заболеваниях. Симптомы со стороны ЖКТ распространены, но недостаточно распознаются у пациентов с митохондриальными заболеваниями; обычно они включают в себя нарушение моторики любого отдела ЖКТ, рефлюксы, дисфункцию глотания, задержку опорожнения желудка, проблемы с кормлением и/или ростом, псевдообструкцию, мальабсорбцию и запоры. Распространенными, но недооцененными являются эндокринные нарушения, включают дисфункцию гипофиза, надпочечников, ЩЖ и ПЖЖ. Такое тщательное обследование пациентов с митохондриальным заболеванием позволяет исключить общие и требующие терапии клинические проявления на момент обследования, хотя не исключено их появление со временем, и наоборот, в случае выявления признаков обеспечивает повышенную диагностическую настороженность и определяет тактику дальнейшего ведения пациента.
в) Наследование митохондриальных заболеваний. Первичное митохондриальное заболевание возникает в результате нарушения генов ядерной и митохондриальной ДНК (мтДНК), которые могут быть унаследованы от родителя или возникать de novo у человека, подвергшегося неблагоприятным воздействиям. Т.о., для митохондриальных заболеваний характерны все модели наследования: по менделевскому (АуР, АуД, Х-сцепленный) или материнскому типу (мтДНК) (табл. 1). Важно получить подробную родословную трех поколений, чтобы потенциально выделить конкретный образец наследования в данной семье.
У пациентов с наследственными нарушениями мтДНК необходимо изучать информацию о членах семьи по материнской линии (как о мужчинах, так и о женщинах, но будут рассматриваться только родственники с нарушениями по женской линии).
Семейный анамнез опирается на наличие функциональных проблем в различных органах, таких как мигрень, усталость, непереносимость физических упражнений, инсульт, СД, дисфункция ЩЖ, СРК, аффективное расстройство или проблемы со зрением и слухом. При Х-сцепленных нарушениях симптомы и тяжелые формы заболевания проявляются у мужчин, в то время как женщины или не имеют нарушений, или они минимальны. АуР-расстройства распространены при детских митохондриальных заболеваниях, особенно при наличии близкородственных браков. В таких случаях редкий в общей популяции вариант становится частым и, передаваясь по материнской и отцовской линиям, становится гомозиготным у пораженного пробанда. Как правило, страдает только одно поколение в родословной. АуД-варианты могут возникать de novo или передаваться от одного из родителей к ребенку, хотя многие нарушения имеют пониженную пенетрантность, что может привести к тому, что при наследовании заболевание будет передаваться через поколение.
Определение вероятногопаттерна наследования с помощью анализа родословной может обеспечить точную интерпретацию крупномасштабных генетических диагностических оценок, таких как мультигенное секвенирование и панели анализа делеции/дублирования, а также секвенирование экзома или генома. Установление правильного генетического диагноза митохондриального заболевания у больного человека имеет большое значение для обеспечения достоверного консультирования по вопросам риска рецидивов. Кроме того, оно необходимо для планирования последующего рождения детей в данных семьях. Варианты пренатальной диагностики с целью выявления конкретного заболевания могут включать в себя исследование ворсинок хориона (CVS, англ. Chorionic villus sampling; обычно выполняется на 10-12-й неделе беременности), амниоцентез (обычно выполняется на сроке беременности 16-20 нед) или предимплантационную генетическую диагностику при проведении ЭКО.
Особенно следует упомянуть об уникальных аспектах материнской наследственности, которые типичны для нарушений мтДНК. В настоящее время выявлено более 300 мутаций мтДНК, а также разл. делеций и перестроек мтДНК, вызывающих заболевание, с большим разнообразием проявлений и особенностей заболевания. Одна из важнейших молекулярно-генетических особенностей заболеваний с митохондриальным наследованием — феномен гетероплазмии.
Мутации мтДНК могут обнаруживаться в состоянии гомоплазмии (наличие только мутантной мтДНК) или гетероплазмии (сочетание мутантной и нормальной мтДНК). Соотношение двух популяций мтДНК, в значительной мере определяющее фенотип, варьирует индивидуально (в т.ч. в/семейно) и в разных тканях одного больного*. Для гетероплазматических вариантов мтДНК точный уровень мутации (процент) может варьироваться в разных тканях человека и может меняться со временем, при этом тяжесть симптомов соответствует разным пороговым уровням мутаций, которые бывает трудно определить и которые обычно различаются в зависимости от органа.
Фоновый набор вариантов фиксированной последовательности генома мтДНК индивида, известный как гаплогруппа, также может влиять на распространенность или тяжесть заболевания мтДНК. В случае идентификации у пациента нового или редкого варианта мутации мтДНК м.б. полезным проведение высокочувствительных методов секвенирования для проверки уровней этой мутации (точность обнаружения при 1% мутаций) с исследованием различных тканей (кровь, моча, слизистые щек, клетки кожи, мышцы), а также проведение исследования в тканях матери пациента или родственников по материнской линии, чтобы точно определить, может ли это быть причиной заболевания в этой семье. Функциональное тестирование, основанное на исследованиях, также м.б. необходимо для полной характеристики эффектов недавно обнаруженного варианта мтДНК.
В случаях, когда неизвестно, наследуется ли вариант мтДНК по материнской линии или возник de novo, риск рецидива для будущего потомства родителя, не имеющего симптомов, эмпирически оценивается в 1 из 25 (4%), а при наличии у матери симптомов эмпирический риск возрастает до 1 из 2 (50%).
P.S. * Руденская Г.Е., Захарова Е.Ю. Наследственные нейрометаболи-ческие болезни юношеского и взрослого возраста. М.: ГЭОТАР-Медиа, 2018.
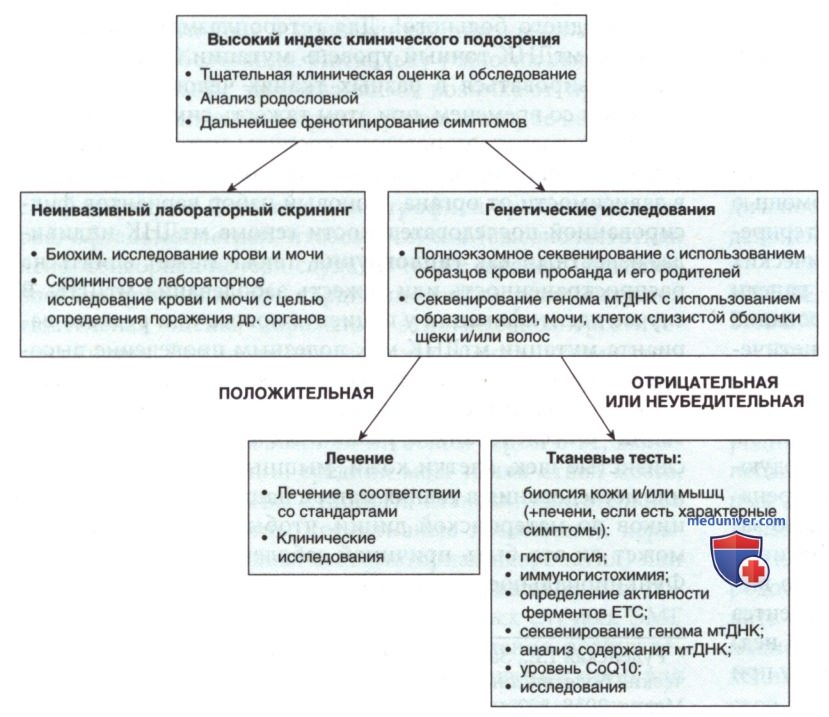
г) Диагностические тесты при митохондриальных заболеваниях. Диагностика митохондриального заболевания основана, прежде всего, на генетическом исследовании (анализе генома). Полезными являются также биохимические анализы крови или мочи. Инвазивное исследование тканей часто рассматривается как вторичное, и иногда проведение его не требуется (рис. 3).
Рисунок 3. Диагностический алгоритм при подозрении на митохондриальное заболевание
Клиническая оценка включает в себя историю болезни — тщательный осмотр органов и систем, физическое, неврологическое обследование и обследование дисморфофобии; биохимические скрининговые исследования на основе родословной, анализов крови и мочи; дополнительные клинические оценки фенотипирования. При подозрении на наличие митохондриального заболевания можно использовать широкий диапазон клинических и диагностических методов исследования. При отсутствии известной молекулярной этиологии у члена семьи, имеющего заболевание, генетическое диагностическое тестирование первой линии может включать в себя целенаправленную панель из сотен или тысяч известных генов ядерной и мтДНК с использованием методологий NGS, с помощью которого можно обнаружить как однонуклеотидные, так и более масштабные делеции и дупликации генов.
Если такое тестирование не дает результатов, м.б. проведено клинически обоснованное секвенирование полного экзома (WES; англ. whole exome sequencing). В настоящее время стандарт оказания МП переходит на использование WES в качестве первоначального диагностического тестирования, которое является более всеобъемлющим для генов, которые, как известно, вызывают не только митохондриальные заболевания, но и все остальные генетические заболевания человека. Обоснованием такой эволюции подхода к диагностическому тестированию являются следующие факторы.
• Все более одинаковая стоимость и время выполнения массовых параллельных исследований NGS на основе панелей и WES.
• Обычная лабораторная практика генетической диагностики — генерировать данные WES для всех заказанных тестов, но оценивать и сообщать только о вариантах в определенных подмножествах генов, когда запрашивается панельное тестирование, оставляя остальные гены без интерпретации.
• При проведении исследования WES крови определение последовательности генома мтДНК часто включается без дополнительных затрат, но может потребоваться повторное исследование пораженных тканей (напр., мышц, печени) для выявления гетероплазматических вариантов мтДНК, которые могут отсутствовать в крови.
• Польза выполнения одновременного секвенирования пробандов и обоих родительских образцов (тестирование на основе трио) проводится с WES, но не делается при панельном тестировании, что позволяет проводить параллельный анализ сегрегации подозреваемых патогенных вариантов, а также легко идентифицировать de novo доминантные варианты у пробанда.
• Клиническо-диагностические лаборатории все чаще сообщают об улучшении диагностической эффективности экзома по сравнению с панельным тестированием, учитывая крайне неоднородную природу митохондриальных заболеваний, быстрые темпы изменений в распознавании новых генетических диагнозов, делающих устаревшими ранее установленные панели генов, и обширное фенотипическое совпадение с немитохондриальными заболеваниями.
• Способность использовать необработанные данные WES (либо на исследовательской основе, либо для повторного анализа позже в клинико-диагностической лаборатории) для определения и/или выявления «новых» генных нарушений, ранее не распознаваемых или не связанных с заболеванием человека.
Ресурс сообщества по митохондриальным заболеваниям, предназначенный для централизованного хранения всех данных о митохондриальных заболеваниях, генах и вариантах обоих геномов, общедоступен на сайте www. mseqdr.org. По оценкам, секвенирование экзома, включая мтДНК, позволяет определить окончательную генетическую этиологию митохондриального заболевания, по крайней мере, у 60% пациентов, у которых предполагается данная патология. Это позволяет сократить «диагностическую одиссею», длящуюся у многих пациентов годы и десятилетия до нескольких месяцев.
Тканевые диагностические тесты стали реже использоваться в качестве первичных тестов у всех пациентов с подозрением на митохондриальные заболевания, несмотря на то что в некоторых случаях они все еще имеют клиническую ценность. К ним относятся тесты 1) в обстановке быстро ухудшающегося клинического статуса, когда результаты генетического тестирования м.б. своевременно недоступны; 2) когда при геномном тестировании идентифицирован вариант с неопределенным значением и имеются неясные биохим. последствия; и 3) когда неинформативное секвенирование генома в крови у пациента с миопатией или мышечными симптомами вызывает предположение о наличии заболеваний, которые могут быть диагностированы при гистологии, электронной микроскопии, иммуногистохимии или исследовании ферментативного состава ткани. Кроме того, некоторые митохондриальные заболевания выявляются только при диагностических исследованиях тканей.
К ним относятся нарушения с делецией мтДНК (обычно с участием нескольких тысяч нуклеотидов), отсутствующие в крови, которые вызывают хроническую прогрессирующую наружную офтальмоплегию или расстройство спектра синдрома Кернса-Сейра, а также различные тканевые (мышцы или печень) специфические расстройства истощения мтДНК (напр., снижение содержания мтДНК в ткани), которые подтверждают митохондриальную патофизиологию у данного пациента и подчеркивают вероятную причину его заболевания из-за нарушения ядерного гена, поскольку для поддержания мтДНК требуется множество ядерно-кодированных белков.
Для комплексной оценки способности окислительного фосфорилирования эндоплазматического ретикулума в мышцах требуется анализ свежей биопсии мышц, и данное исследование доступно только в очень ограниченном количестве исследовательских центров в мире. Признанным золотым стандартом для оценки митохондриальной дисфункции является анализ активности ферментов цепи переноса электронов в ранее замороженном образце ткани, он может выполняться в любом медучреждении. С целью исследования функции митохондрий в культуре фибробластов м.б. выполнена биопсия кожи. При выявлении нарушений м.б. указан конкретный тип митохондриальной болезни, хотя не все митохондриальные заболевания могут обнаруживаться при анализе кожи. Т.о., если заболевание не было выявлено при тестировании фибробластов, впоследствии может потребоваться проведение более инвазивных исследований тканей.
Исследование клеточных линий фибробластов и лимфоцитов можно отнести к малоинвазивным методам, при этом обладающим высокой диагностической ценностью, позволяющим проводить анализ активности ферментов клеток, валидацию генов и на основании полученных результатов подбирать терапию.
д) Принципы лечения митохондриального заболевания. Отсутствуют эффективные методы лечения как первичных, так и вторичных митохондриальных заболеваний, поскольку недостаточно данных о биохимических и физиол. изменениях, которые способствуют их разнообразным клиническим проявлениям. Клиническая сложность и неточно определенные или недостаточно понятные биохим. фенотипы разл. подтипов митохондриальных заболеваний затрудняют для клиницистов эффективное применение или мониторинг таргетной терапии при заболеваниях ДЦ. Митохондриальные коктейли из витаминов и добавок могут включать витамины [В1, В2, аскорбиновая кислота («Витамин С»)], антиоксиданты [коэнзим Q10, тиоктовая кислота («Липоевая кислота»), витамин Е] и модификаторы метаболизма (креатин, L-карнитин, L-аргинин, фолиевая кислота).
Хотя эффективность, токсичность и оптимальная доза этих ЛП неизвестны и не имеют объективной оценки применения у пациентов с болезнью ДЦ, их продолжают назначать эмпирически в целях улучшения остаточной ферментативной функции ДЦ или подавления токсических метаболитов, которые, как предполагается, накапливаются при ДЦ-дисфункции, а также в связи с сообщениями пациентов об улучшении самочувствия. При назначении этих методов лечения используется универсальный подход, при котором не учитываются варианты подтипов первичных митохондриальных заболеваний, тканеспецифические проявления и основные патогенные факторы, такие как преобладающие метаболические и сигнальные изменения, характерные для разл. подклассов заболеваний.
Хотя на сегодняшний день для митохондриальных заболеваний не существует ЛП или терапии, одобренной FDA, усовершенствованное молекулярное разграничение позволило избранным методам лечения перейти от теоретической, эмпирической и в значительной степени неэффективной стадии к многообещающему горизонту рационального, персонализированного и эффективного вмешательства. Все большее число диагнозов митохондриальных заболеваний требует лечения, включающего назначение или отказ от приема специфических ЛС (ГКС, вальпроевая кислота, фенитоин, барбитураты, пропофол в течение длительного периода времени, превышающее 30-60 мин, некоторые анестетики, статины, β-блокаторы, амиодарон, нуклеозидные ингибиторы обратной транскриптазы), обеспечения кофакторами или диетами, а также своевременного скрининга возможных клинических проявлений. Общие методы лечения синдрома Лея, такие как назначение L-аргинина и цитруллина, могут предотвратить или остановить развитие последствий метаболического инсульта для НС.
Нутриционная терапия при этих расстройствах адаптирована к конкретным генетическим заболеваниям. Так, тиамин и биотин используются при болезни SLC19A3, убихинрл для болезни PDSS2 (дефицит коэнзима Q10), тиамин и кетогенная диета для дефицита PDHA1 (англ. pyruvate dehydrogenase —пируватдегидрогеназы). Точный молекулярный диагноз может также спасти жизнь за счет отказа от голодания, отмены приема митохондриально-токсичных ЛС или использования общих анестетиков при определенных подгруппах митохондриальных заболеваний. Кроме того, это позволяет проводить более полноценные консультации, провести профилактику развития рецидивов, обеспечить скрининг мед. осложнений и в некоторых случаях — использовать необходимые кофакторы или витамины.
Кроме того, в некоторых странах для профилактики наследственных вариантов мтДНК используются репродуктивные технологии, такие как технологии заместительной митохондриальной терапиипри ЭКО. Наконец, возможность молекулярной идентификации пациентов с первичными митохондриальными заболеваниями позволила разработать все большее количество клинических экспериментальных методов лечения, которые в настоящее время планируются или проводятся в отношении разнообразного спектра симптомов, возникающих при первичных митохондриальных заболеваниях.