Роль гликогена в мышцах заключается в обеспечении субстрата для выработки АТФ для сокращения мышц. БНГ мышц можно разделить на две группы. Первая группа характеризуется гипертрофической кардиомиопатией, нарастающей слабостью и атрофией скелетных мышц или и тем и другим и связана с недостаточностью кислой а-глюкозидазы, лизосомального фермента, разрушающего гликоген (БНГ типа II), LAMP2 и PRKAG2.
Вторая группа, связанная с нарушением энергетического обмена в мышцах, характеризуется мышечной болью, непереносимостью физических упражнений, миоглобинурией и быстрой утомляемостью. К этой группе относятся недостаточность мышечной фосфорилазы (болезнь Мак-Ардла, БНГ типа V) и недостаточность мышечной фосфофруктокиназы (типа VII, болезнь Таури), фосфоглицераткиназы, фосфоглицератмутазы, лактатдегидрогеназы и мышечно-специфической киназы фосфорилазы. При недостаточности некоторых из этих ферментов наблюдается компенсированный гемолизом, что указывает на генерализованные нарушения метаболизма глюкозы.
а) Гликогеновая болезнь II (дефицит а-1,4-глюкозидазы, болезнь Помпе; болезнь накопления гликогена II типа; дефицит кислой мальтазы; гликогеноз II типа)*.
P.S. * КР «Болезнь Помпе», М3 РФ, 2019 г.
Болезнь Помпе, также называемая БНГ типа II или недостаточностью кислой мальтазы, вызвана дефицитом кислой α-1,4-глюкозидазы (кислой мальтазы) — фермента, ответственного за расщепление гликогена в лизосомах. Это приводит к накоплению лизосомального гликогена во многих тканях и типах клеток, преимущественно затрагивая скелетные, сердечные и гладкомышечные клетки. При болезни Помпе гликоген обычно накапливается в лизосомах, в отличие от его накопления в цитоплазме при др. гликогенозах. Однако по мере прогрессирования болезни происходит разрыв лизосом, утечка из них, что приводит к появлению цитоплазматического гликогена.
Болезнь Помпе — АуР-заболевание. Считалось, что заболеваемость составляет ~1:40 ООО живорождений у кавказцев и 1:18 000 живорождений у китайских ханьцев. Скрининг новорожденных на болезнь Помпе в США показывает, что распространенность намного выше, чем предполагалось ранее (от 1:9132 до 1:24 188). Ген кислой α-глюкозидазы (GAA; англ, glucosidase alpha acid) находится на хромосоме 17q25.2. Было идентифицировано >500 патогенных вариантов, которые м.б. использованы для определения фенотипов. При взрослой форме болезни обнаруживается сплайсинг РНК (IVS1-13T→G; c.-32-13T>G), обычно наблюдается у пациентов европеоидной расы.
1. Клинические проявления. С учетом единого патогенеза болезни Помпе, выделяют только два варианта в зависимости от времени манифеста симптомов: младенческую (инфантильную), манифестирующую в период новорожденности или младенческом возрасте, и болезнь Помпе с поздним началом.
Младенческая болезнь Помпе всегда приводит к летальному исходу без ферментозамещающей терапии (ФЗТ) с алглюкозидазой альфа*. У больных новорожденных с первых дней жизни появляется гипотония, генерализованная мышечная слабость — типичный вид «вялого ребенка», невропатическая бульбарная слабость, вялое сосание, макроглоссия, гепатомегалия и гипертрофическая кардиомиопатия, которые при отсутствии лечения приводят к смерти от кардиореспираторной недостаточности или респираторной инфекции, обычно к 1-му году жизни.
Болезнь Помпе с поздним началом (ювенильное, детское и взрослое заболевание*) характеризуется слабостью проксимальных мышц конечностей и ранним поражением дыхательных мышц, особенно диафрагмы. Поражение сердца варьируется от нарушений сердечного ритма до кардиомиопатии и менее тяжелых краткосрочных прогнозов. Симптомы, связанные с прогрессирующей дисфункцией скелетных мышц, могут проявиться в возрасте с 1 года до 60 лет. В клинической картине преобладает постепенно нарастающая слабость проксимальных групп мышц, в том числе мышц туловища. Более сильно поражаются нижние конечности, чем верхние.
Особенно сильно страдают мышцы тазового пояса и спины, а также диафрагма. Др. симптомы могут включать слабость языка, птоз и расширение кровеносных сосудов (напр., базилярной артерии, восходящей аорты). По мере прогрессирования заболевания пациенты становятся прикованы к инвалидной коляске и нуждаются в ИВЛ. Первоначальными симптомами у некоторых пациентов м.б. дыхательная недостаточность, проявляющаяся сонливостью, утренней головной болью, ортопноэ и одышкой при ФН, которые в конечном счете приводят к нарушению дыхания во сне и ДН. ДН является причиной значительной заболеваемости и смертности при болезни Помпе с поздним началом.
Аневризмы базилярной артерии с разрывом также в некоторых случаях способствуют летальному исходу. У некоторых пациентов с болезнью Помпе с поздним началом была выявлена невропатия мелких волокон, проявляющаяся как болезненная парестезия. Сообщалось о желудочно-кишечных расстройствах, таких как вздутие живота после приема пищи, дисфагия, раннее насыщение, диарея, хронические запоры и СРК. Поражение МВП не является редкостью и может проявляться недержанием мочевого пузыря и кишечника, слабой струей мочи или подтеканием.
При отсутствии лечения возраст смерти варьируется от раннего детства до позднего взросления, в зависимости от скорости прогрессирования заболевания и степени поражения дыхательных мышц. С появлением ФЗТ возникают новые исходы и прогнозы как для детей, имеющих младенческую форму, так и для болезни Помпе с поздним началом.
P.S. * В РФ в настоящий момент эти выделения не используют См. КР «Болезнь Помпе».
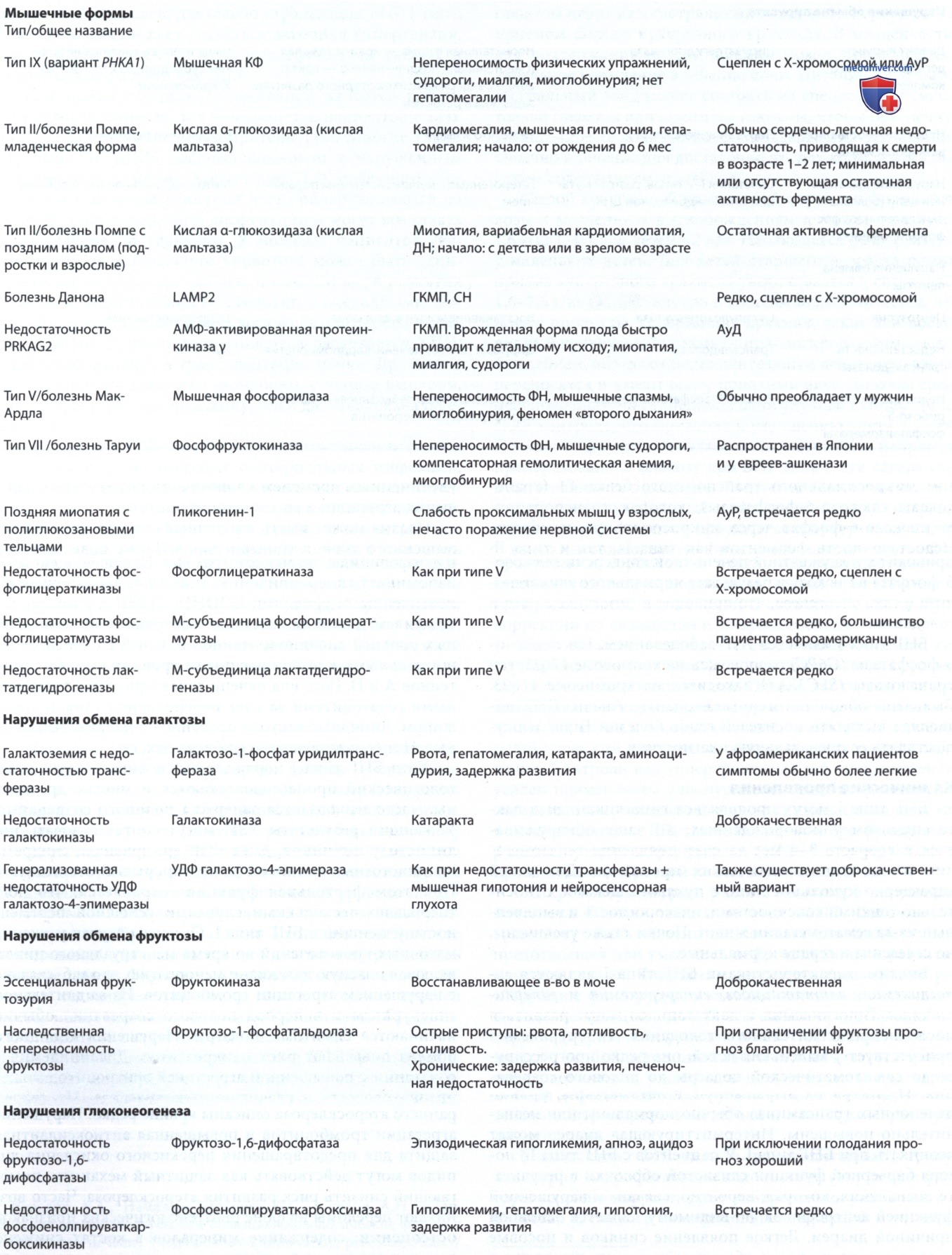
2. Лабораторные признаки. К ним относятся повышенные уровни в сыворотке КФК, ACT, АЛТ и ЛДГ. Тетрасахарид глюкозы в моче, метаболит разложения гликогена, является надежным биомаркером тяжести заболевания и реакции на лечение. При младенческой форме часто первым симптомом оказывается массивная кардиомегалия при рентгенологическом исследовании органов грудной клетки. Данные ЭКГ включают высоковольтный комплекс QRS, синдром WPW и укороченный интервал Р-R. ЭхоКГ выявляет утолщение обоих желудочков, и/или МЖП, и/или обструкции выводящего тракта ЛЖ. В биоптатах мышц обнаруживают вакуоли с содержимым, которые положительно окрашиваются на гликоген. Повышена кислая фосфатаза, предположительно из-за компенсаторного увеличения лизосомальных ферментов. Электронная микроскопия выявляет накопление гликогена в везикулах и в цитоплазме.
ЭМГ выявляет признаки миопатии с повышенной электрической возбудимостью мышечных волокон и псевдомио-тоническими разрядами. У взрослых пациентов уровень сывороточной КФК повышен не всегда. В зависимости от взятой или протестированной мышцы гист. вид мышц и ЭМГ м.б. нормальными. Необходимо тестировать именно пораженные мышцы.
Некоторые пациенты с детской болезнью Помпе, которым была сделана биопсия периферических нервов, продемонстрировали накопление гликогена в нейронах и шванновских клетках.
3. Диагностика. Диагноз болезни Помпе м.б. поставлен с помощью ферментного анализа на сухих пятнах крови, лейкоцитах, мононуклеарных клетках крови, мышцах или культуре кожных фибробластов, демонстрирующих снижение активности кислой а-глюкозидазы. Подтверждением является секвенирование гена, показывающее два патогенных варианта в гене GAA. Ферментный анализ следует проводить в лаборатории с практикой использования мальтозы, гликогена или 4-метилумбеллиферил-α-D-глюкопиранозида (4MUG; англ. 4-Methylumbelliferyl-α-D-glucopyranoside) в качестве субстрата.
Младенческая форма отличается более серьезным дефицитом ферментов, чем формы с поздним началом. Определение процента остаточной активности фермента фиксируется в фибробластах кожи и мышцах. Анализы крови, особенно сухих пятен крови, обладают преимуществом быстрого анализа и все чаще используются в качестве первичного материала для постановки диагноза. Биопсия мышцы часто проводится при подозрении на мышечное заболевание и для ДД; он дает более быстрые результаты и предоставляет дополнительную информацию о содержании гликогена и месте хранения гликогена внутри и вне лизосом мышечных клеток.
Однако нормальные показатели при биопсии мышц не исключают диагноза болезни Помпе. У пациентов с поздним началом заболевания наблюдается вариабельность накопления гликогена в различных мышцах и внутри мышечных волокон; гистология мышц и содержание гликогена могут варьироваться в зависимости от места биопсии мышцы. Также существует высокий риск опасности при проведении анестезии у младенцев. ЭКГ полезна при постановке диагноза при подозрении на младенческую форму и должна проводиться пациентам с подозрением на болезнь Помпе до проведения любой процедуры, требующей анестезии, включая биопсию мышц.
В моче пациентов м.б. повышены тетрасахариды глюкозы, их уровни чрезвычайно высоки у детей младшего возраста. Наличие панелей секвенирования нового поколения и всего экзома позволяет идентифицировать дополнительных пациентов с болезнью Помпе, особенно когда диагноз неоднозначен. Доступна пренатальная диагностика с использованием амниоцитов или ворсинок хориона.
4. Лечение. Для лечения болезни Помпе доступна ФЗТ рекомбинантной кислой α-глюкозидазой человека (аглюкозидаза альфа). Рекомбинантная кислотная α-глюкозидаза восполняет активность лизосомальной кислой альфаглюкозидазы, что приводит к стабилизации или восстановлению функции сердечной и скелетных мышц (включая дыхательные мышцы) (рис. ниже). ФЗТ следует начинать как можно раньше при всех формах заболевания, особенно у новорожденных с младенческой формой, потому что болезнь быстро прогрессирует. Дети, у которых отсутствует перекрестно реагирующий иммунологический материал, вырабатывают АТл с высоким титром против введенного фермента, и они менее благоприятно реагируют на ФЗТ.
Рентгенография органов грудной клетки и данные гистологии мышц пациента с младенческой болезнью Помпе до (А) и после (В) ферментозамещающей терапии. Обратите внимание на уменьшение размера сердца и гликогена в мышцах на фоне терапии.
Лечение с использованием иммуномодулирующих агентов, таких как метотрексат, ритуксимаб и в/в иммуноглобулин, продемонстрировало эффективность в предотвращении развития иммунного ответа на ФЗТ и иммунной толерантности. При наличии показаний следует использовать ночную ИВЛ; было показано, что это улучшает качество жизни и особенно полезно в период декомпенсации дыхания.
Помимо ФЗТ, у пациентов с болезнью Помпе были продемонстрированы преимущества др. дополнительных методов лечения. Для пациентов с поздним началом болезни м.б. полезна диета с высоким содержанием белка. Силовые тренировки дыхательных мышц показали улучшение респираторных параметров в сочетании с ФЗТ. Субмаксимальные режимы упражнений помогают улучшить мышечную силу, уменьшить боль и утомляемость. Др. методы находятся в стадии клинической разработки и направлены на повышение безопасности и эффективности доставки ферментов к пораженным тканям.
К ним относятся использование молекул шаперонов для усиления доставки rhGAA и neoGAA, что представляет собой ФЗТ второго поколения с большим количеством тегов маннозо-6-фосфата (М6Р; англ. Mannose-6-phosphate), которые усиливают нацеливание на рецепторы М6Р и захват ферментов. Исследования генной терапии для коррекции путей выработки эндогенных ферментов также являются многообещающими.
Ранняя диагностика и лечение необходимы для достижения оптимальных результатов. Скрининг новорожденных с использованием анализов крови на Тайване привел к раннему выявлению случаев заболевания Помпе и, таким образом, к улучшению исходов болезни за счет раннего начала ФЗТ.
б) Заболевания накопления гликогена, имитирующие гипертрофическую кардиомиопатию (болезнь Данона). Причиной болезни Данона является мутация гена LAMP2 и, как следствие, дефицит LAMP2 — важного компонента лизосомальной мембраны. Это приводит к накоплению гликогена в сердце и скелетных мышцах, что в первую очередь проявляется ГКМП и слабостью скелетных мышц. Болезнь Данона можно отличить от обычных причин ГКМП (дефекты генов саркомерных белков) по аномалиям проводящих путей сердца, особенно по предвозбуждению желудочков и дефектам проводимости. Пациенты обращаются с сердечными симптомами, включая боль в груди, учащенное сердцебиение, синкопальные состояния и перебои в сердце, обычно в возрасте от 8 до 15 лет.
Др. клинические проявления болезни Данона включают периферическую пигментную ретинопатию, изменения хрусталика и аномальные электроретинограммы. Это заболевание наследуется по сцепленному с Х-хромосомой доминантному типу. Диагноз можно поставить путем генетического тестирования гена LAMP2. Прогноз дефицита LAMP2 неблагоприятный, что связано с прогрессирующей сердечной недостаточностью в терминальной стадии в раннем возрасте.
Описана триада симптомов болезни Данона: кардиомиопатия, скелетная миопатия (периферические мышечные нарушения) и отставание в умственном развитии. Вместе с тем у ряда пациентов описана минимальная выраженность мышечных и когнитивных нарушений1.
Лечение направлено на сдерживание симптомов у пораженных людей, включая лечение кардиомиопатии, коррекцию аритмий и физиотерапию при мышечной слабости. Некоторым пациентам была успешно проведена трансплантация сердца.
в) Недостаточность аденозинмонофосфат-активированной протеинкиназы γ2 (недостаточность АМФ-активированной протеинкиназы γ2). Недостаточность PRKAG2 вызвана патогенными вариантами гена PRKAG2, картированного на хромосоме 7q36. PRKAG2 необходим для синтеза фермента АМФ-активируемой протеинкиназы (АМРК; англ. AMP-activated protein kinase), которая регулирует клеточные пути, участвующие в метаболизме АТФ. Общие проявления включают ГКМП и аномалии проводящих путей, такие как синдром WPW, ФП и прогрессирующая АВ-блокада. Поражение сердца варьирует и включает наджелудочковую тахикардию, синусовую брадикардию, дисфункцию ЛЖ и даже в некоторых случаях внезапную сердечную смерть.
Помимо поражения сердца, существует широкий спектр фенотипических проявлений, включая миалгию, миопатию и судороги. Кардиомиопатия, вызванная вариантами PRKAG2, обычно обеспечивает долгосрочное выживание, хотя редкая врожденная форма, проявляющаяся в раннем младенчестве, связана с быстрым летальным исходом. Кардиомиопатия при синдроме PRKAG2 часто имитирует кардиомиопатию при др. состояниях, особенно при болезни Помпе, ее следует рассматривать как ДД у младенцев с тяжелой ГКМП. Лечение в основном симптоматическое, включая лечение СН и коррекцию дефектов проводимости.
г) Мышечный гликогеноз. Этот гликогеноз является результатом недостаточности мышечной гликогенсинтазы [гликогенсинтазы I (GYS1; англ. glycogen synthase 1)]. Ген GYS1 был локализован на хромосоме 19q13.3. В прямом смысле это не тип гликогеноза, потому что дефицит фермента приводит к уменьшению запасов гликогена. Заболевание встречается крайне редко и было зарегистрировано у троих детей единокровных родителей сирийского происхождения. Биопсия мышц показала отсутствие гликогена, преимущественно окислительных волокон, и митохондриальную пролиферацию. Толерантность к глюкозе была нормальной. Молекулярное исследование выявило гомозиготную стоп-мутацию (R462→ter) в гене мышечной гликогенсинтазы.
Фенотип был разным у трех братьев и сестер и варьировался от внезапной остановки сердца, мышечной утомляемости, ГКМП, аномальной частоты сердечных сокращений и гипотонии во время физических упражнений до легкого нарушения сердечной функции в покое.
д) Поздняя полиглюкозановая миопатия тела (из вариантов GYG1). Поздняя полиглюкозановая миопатия тела — это АуР, медленно прогрессирующая скелетная миопатия, вызванная патогенными вариантами гена GYG1, блокирующими биосинтез гликогенина-1. Гликогенин-1, который является предшественником, необходимым для образования гликогена, снижен или полностью отсутствует. Накопление полиглюкозана в скелетных мышцах вызывает у взрослых слабость проксимальных групп мышц, в основном поражаются мышцы тазобедренного и плечевого пояса. Поражения сердца не наблюдается. По сравнению с БНГ типа IV — болезнью полиглюкозановых телец у взрослых, — вовлечение НС встречается редко, хотя отложение полиглюкозана наблюдается при обоих расстройствах.
GYG1 отображается на хромосоме 3q24. При биопсии мышц 30-40% мышечных волокон имеют положительную ШИФ-реакцию в связи с накоплением полиглюкозана. Электронная микроскопия показывает типичную структуру полиглюкозана овоидной формы, состоящую из частично нитевидного материала.
е) Гликогеновая болезнь типа V (гликогеноз типа V, дефицит мышечной фосфорилазы, болезнь Мак-Ардла). БНГ типа V вызывается недостаточностью активности мышечной фосфорилазы. Недостаток этого фермента ограничивает выработку мышечного АТФ за счет гликогенолиза, что приводит к накоплению мышечного гликогена и является прототипом нарушения энергетического обмена в мышцах. Недостаточность мышечной фосфорилазы нарушает отщепление молекул глюкозила от прямой цепи гликогена.
1. Клинические проявления. Симптомы обычно впервые появляются в позднем детстве или в течение второго десятилетия жизни. Гетерогенность клинической картины встречается редко, но описаны и случаи, свидетельствующие об обратном. Исследования показали, что болезнь Мак-Ардла может проявляться как у людей в возрасте от 74 лет, так и в периоде новорожденности в ранней форме с летальным исходом с гипотонией, общей мышечной слабостью и прогрессирующей ДН. Симптомы обычно характеризуются непереносимостью физических упражнений с мышечными спазмами и болью. Симптомы проявляются при двух типах активности: упражнениями высокой интенсивности, такими как бег на короткие дистанции или перенос тяжелых грузов, и менее интенсивными, но продолжительными действиями, такими как подъем по лестнице или ходьба в гору.
Большинство пациентов хорошо переносят умеренные упражнения в течение длительного времени, такие как ходьба по ровной поверхности. Многие пациенты испытывают характерный феномен «второго дыхания», когда мышечные боли и усталость проходят после короткого отдыха. Пациенты с данным типом миопатии могут подвергаться риску статин-индуцированной миопатии и рабдомиолизу. Обычно пациенты испытывают эпизодические мышечные боли и спазмы от упражнений, но 35% пациентов с болезнью Мак-Ардла имели жалобы на постоянные боли, которые серьезно влияли на сон и др. виды деятельности. Исследования также предполагают, что м.б. связь между БНГ типа V и разл. когнитивными нарушениями.
Приблизительно 50% пациентов сообщают о бордовом цвете мочи после тренировки в результате миоглобинурии, вызванной ФН, и рабдомиолиза. Чрезмерная миоглобинурия после интенсивных упражнений может спровоцировать ОПН.
Результаты лабораторных исследований показывают повышенный уровень КФК в сыворотке крови в состоянии покоя, который еще больше увеличивается после тренировки. Физические упражнения также повышают уровень аммиака, инозина, гипоксантина и мочевой кислоты в крови, что м.б. связано с ускоренной рециркуляцией пуриновых нуклеотидов в мышцах, вызванной недостаточной выработкой АТФ. БНГ типа V является АуР-заболеванием. Ген мышечной фосфорилазы (PYGM; англ. glycogen phosphorylase, muscle associated) расположен на хромосоме 11q13.
2. Диагностика. Стандартный диагноз «БНГ типа V» включает биопсию мышц для измерения содержания гликогена, а также активности фермента и секвенирования PYGM. Метаболическую миопатию можно быстро диагностировать путем проведения ишемической пробы с ФН. На мышечный гликогеноз, связанный с дефектом превращения мышечного гликогена или глюкозы в молочную кислоту, указывает отсутствие прироста уровня лактата и чрезмерное повышение уровня аммиака в крови. Аномальная ишемическая реакция на ФН не ограничивается БНГ типа V. Др. мышечные дефекты гликогенолиза или гликолиза дают аналогичные результаты (недостаточность мышечной фосфофруктокиназы, фосфоглицераткиназы, фосфоглицератмутазы или ЛДГ).
Ишемическая проба с нагрузкой когда-то использовалась в качестве быстрого диагностического скрининга для пациентов с подозрением на заболевание, но была связана с серьезными осложнениями и л/п-результатами. Было установлено, что неишемический тест с ФН для предплечья обладает высокой чувствительностью, его легко выполнять, он является экономически эффективным и свидетельствует о гликогенозе мышц. Однако, как и в случае с ишемическим тестом, он не может отличить аномальные реакции на упражнения, вызванные заболеванием V типа, от др. дефектов гликогенолиза или гликолиза или фермента ветвления (отмечается, когда тест проводится после голодания).
Диагноз подтверждается молекулярно-генетическим тестированием PYGM. Распространенный нонсенс-вариант, p.R49X в экзоне 1, обнаруживается у 90% пациентов европеоидной расы, а делеция одного кодона в экзоне 17 — у 61% пациентов из Японии. Вариант p.R49X представляет 55% аллелей у испанских пациентов, тогда как вариант p.W797R — 14%, a p.G204S — 9% патогенных аллелей в популяции Испании. По-видимому, существует связь между клинической тяжестью БНГ типа V и наличием аллеля D полиморфизма инсерции/делеции АПФ. Это может объяснить спектр фенотипической изменчивости, проявляющейся при этом заболевании.
3. Лечение. Клинические симптомы можно предотвратить, исключив чрезмерные ФН. Рекомендуются регулярные и умеренные упражнения для улучшения работоспособности мышц. Глюкоза или сахароза, вводимые перед тренировкой или инъекцией глюкагона, также могут значительно увеличить переносимость нагрузки у этих пациентов. Диета с высоким содержанием белка может повысить физическую выносливость, а прием низких доз креатина улучшает функцию мышц у некоторых пациентов. Клинический ответ на креатин зависит от дозы; мышечная боль может усиливаться при приеме высоких доз креатина. Добавка витамина В6 снижает непереносимость ФН и мышечные судороги. На продолжительность жизни это заболевание обычно не влияет.
ж) Гликогеновая болезнь типа VII (недостаточность мышечной фосфофруктокиназы, болезнь Таруи). БНГ типа VII вызывается патогенными вариантами гена PFKM, расположенного на хромосоме 12q13.1, что приводит к дефициту фермента фосфофруктокиназы мышечного типа. Этот фермент является ключевым регуляторным ферментом гликолиза и необходим для АТФ-зависимого превращения фруктозо-6-фосфата во фруктозо-1,6-дифосфат. Фосфофруктокиназа состоит из трех субъединиц изофермента в соответствии с типом ткани и кодируется разными генами: PFKM (М: мышца), PFKL (L: печень) и PFKP (Р: тромбоцит). Скелетные мышцы имеют только субъединицу М, тогда как эритроциты экспрессируют гибрид форм L и М. При БНГ типа VII изофермент М является дефектным, что приводит к полному дефициту активности фермента в мышцах и частичному дефициту в эритроцитах.
БНГ типа VII является АуР-заболеванием, которое чаще встречается у японцев и евреев ашкенази. Дефект сплайсинга и делеция нуклеотида в PFKM составляют 95% патогенных вариантов у евреев ашкенази. Т.о., в этой популяции возможен диагноз на основе молекулярного тестирования типичных вариантов.
1. Клинические проявления. Хотя клиническая картина аналогична клинической картине БНГ типа V, отличительными особенностями БНГ типа VII являются следующие:
• непереносимость ФН, которая обычно начинается в детстве, более серьезна, чем при болезни типа V, и может сопровождаться тошнотой, рвотой и сильной мышечной болью; энергичная нагрузка вызывает сильные мышечные спазмы и миоглобинурию;
• возникает гемолиз разной степени тяжести, на что указывает повышенный уровень билирубина в сыворотке крови и повышенное количество ретикулоцитов;
• гиперурикемия является обычным явлением и усиливается при нагрузке на мышцы в большей степени, чем при БНГ типа V или III;
• в мышечных волокнах присутствует аномальный полисахарид; он дает «+» ШИК-реакцию, но устойчив к амилазе;
• непереносимость физических упражнений особенно усиливается после приема пищи, богатой углеводами, поскольку глюкоза, которая не может утилизироваться мышцами, ингибирует липолиз и тем самым лишает мышцы других субстратов окисления — жирных кислот и кетоновых тел. Это отличается от пациентов с заболеванием типа V, которые могут метаболизировать переносимую с кровью глюкозу, полученную либо в результате эндогенного гликогенолиза печени, либо из экзогенной глюкозы; инфузия глюкозы улучшает переносимость ФН у пациентов типа V;
• феномен «второго дыхания» отсутствует из-за неспособности расщеплять глюкозу в крови.
Встречается несколько редких вариантов БНГ типа VII. Один вариант проявляется у грудных детей в виде мышечной гипотонии и слабости конечностей и переходит в быстро прогрессирующую миопатию, которая приводит к смерти к 4 годам. Второй вариант возникает в младенчестве и приводит к врожденной миопатии и артрогрипозу с летальным исходом. Третий вариант проявляется у грудных детей гипотонией, легкой задержкой развития и судорогами. Дополнительным проявлением является наследственная несфероцитарная гемолитическая анемия. Хотя эти пациенты не имеют мышечной симптоматики, остается неясным, разовьются ли эти симптомы в течение их дальнейшей жизни.
Еще один вариант наблюдается у взрослых и характеризуется медленно прогрессирующей мышечной слабостью, не сопровождается судорогами и миоглобинурией. Также вследствие накопления гликогена может сформироваться утолщение створок митрального клапана.
2. Диагностика. Для постановки диагноза требуются биохимические или гистохимические доказательства дефицита фермента в мышцах. Отсутствие М-субъединицы фосфофруктокиназы также может наблюдаться в мышцах, клетках крови и фибробластах. Секвенирование гена может идентифицировать патогенные варианты гена фосфофруктокиназы.
3. Лечение. Специфическое лечение отсутствует. Для предотвращения приступов мышечных спазмов и миоглобинурии следует избегать интенсивной нагрузки. Употребление простых углеводов перед ФН может улучшить их переносимость. Сообщалось, что кетогенная диета показывает клиническое улучшение у пациента с инфантильной формой БНГ типа VII. Следует избегать таких ЛС, как статины. Необходимо принимать меры предосторожности при проведении наркоза во избежании гипертермии. Употребление углеводов и р-ра глюкозы в пище может привести к ухудшению симптомов из-за неспособности организма усваивать глюкозу. Введенная глюкоза снижает уровень жирных кислот в крови, основного источника мышечной энергии.
з) Мышечно-специфическая недостаточность киназы фосфорилазы (из вариантов РНКА1). Известно несколько случаев дефицита КФ, ограниченного мышцами. Пациенты, как мужчины, так и женщины, жалуются либо на мышечные судороги и миоглобинурию при ФН, либо на прогрессирующую мышечную слабость и атрофию. Активность КФ снижена в мышцах, но остается нормальной в печени и клетках крови. Гепатомегалии или кардиомегалии нет. Тип наследования — сцепленный с Х-хромосомой или АуР. Ген мышечно-специфической формы α-субъединицы (аМ) расположен в Xq12. Патогенные варианты гена были обнаружены у некоторых пациентов мужского пола с этим заболеванием. Ген мышечной γ-субъединицы (γМ, PHKG1) находится на хромосоме 7p12. О патогенных вариантах этого гена пока не сообщалось.
и) Другие мышечные гликогенозы с нарушением энергетического обмена в мышцах. Шесть дополнительных дефектов ферментов — фосфоглицераткиназа, фосфоглицератмутаза, лактатдегидрогеназа, фруктозо-1,6-бисфосфатальдолаза А, мышечная пируваткиназа и β-енолаза в пути терминального гликолиза — вызывают симптомы и признаки энергетической недостаточности, аналогичные БНГ типов V и VII. Неспособность повышать уровень молочной кислоты в крови в ответ на ФН является полезным диагностическим тестом и может использоваться для дифференциации мышечного гликогеноза от нарушений липидного обмена, таких как недостаточность карнитинпальмитоилтрансферазы II и ацил-КоА-дегидрогеназы с очень длинной цепью, которые также вызывают мышечные судороги и миоглобинурию.
При нарушении конечных этапов гликолиза уровень гликогена в мышцах может оставаться нормальным, и для постановки окончательного диагноза необходимо определение активности ферментов в мышечной ткани. Специального лечения не существует.