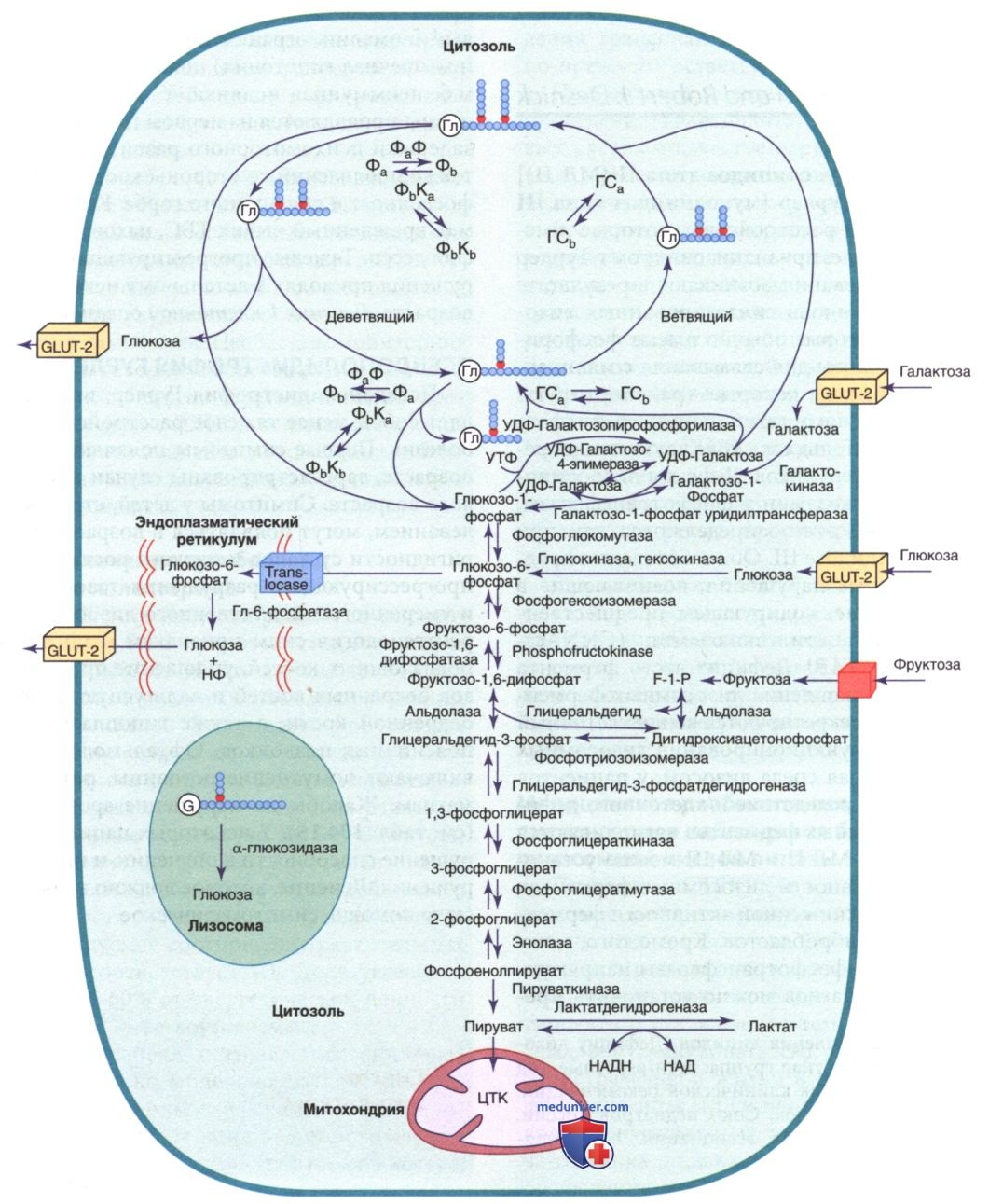
Нарушения метаболизма гликогена, гликогеновая болезнь (гликогенозы, болезни накопления гликогена) являются результатом дефицита разл. ферментов или транспортных белков на путях метаболизма гликогена (см. рис. 1). При этом может изменяться как количество гликогена, так и его качество или и то и другое. Болезни накопления гликогена (БНГ) классифицируются по числовому типу в соответствии с хронологическим порядком, в котором были идентифицированы эти ферментативные дефекты.
Такая числовая классификация до сих пор широко используется, по крайней мере, до номера VII.
В зависимости от локализации дефицитного фермента выделяют печеночную и мышечную форму заболевания*.
P.S. * МЗ РФ. КР «Гликогеновая болезнь у детей», 2016 г.
Нарушение расщепления гликогена может поражать в первую очередь печень и/или мышцы, поэтому в настоящее время оправданным также считается патогенетическое деление БНГ на печеночные, мышечные и смешанные формы*.
P.S. * Баранов А.А., Намазова-Баранова Л.С., Сурков А.Н. и др. Ведение детей с гликогеновой болезнью (нозологические формы с поражением печени). Современные клинические рекомендации // Педиатрическая фармакология. 2020. Т. 17. № 4. С. 303-317.
Сейчас принято выделять 12 (15**) разл. типов ГБ. В раннем детстве проявляются ГБ, связанные с недостаточностью глюкозо-6-фосфатазы (I типа), дефицитом кислой мальтазы (кислой альфа-глюкозидазы) в лизосомах (II типа)***, дефицитом гликоген-деветвящего фермента (III типа) и киназы фосфорилазы печени (IX типа). В подростковом и зрелом возрасте проявляется дефицит мышечной фосфорилазы (V типа, болезнь Мак-Ардля). Кумулятивная частота случаев всех форм БНГ ~1:20 000 живорождений.
P.S. ** МЗ РФ. КР «Гликогеновая болезнь у детей», 2016 г.
Баранов А.А., Намазова-Баранова Л.С., Сурков А.Н. и др. Ведение детей с гликогеновой болезнью (нозологические формы с поражением печени). Современные клинические рекомендации // Педиатрическая фармакология. 2020. Т. 17. № 4. С. 303-317.
P.S. *** Болезнь Помпе. КР, 2019 г. Разработчик клинической рекомендации: Ассоциация медицинских генетиков, Союз педиатров России, РОО «Общество специалистов по нервно-мышечным заболеваниям». Одобрено Научно-практическим советом МЗ РФ.
Общая ориентировочная численность детей с гликогеновой болезнью на территории РФ составляет 0,34:100 000.
Гликогенозы, которые в основном влияют на печень, включают недостаточность глюкозо-6-фосфатазы (типа I), дефицит гликоген-деветвящего фермента (типа III), дефицит гликоген-ветвящего фермента (типа IV), фосфорилазы печени (типа VI), дефект фермента киназы фосфорилазы (типа IX, ранее гликогеноз VIa), недостаточность гликогенсинтазы (типа 0) и дефект транспортера глюко-зы-2. Поскольку метаболизм углеводов в печени отвечает за гомеостаз глюкозы в плазме, эта группа нарушений обычно вызывает гипогликемию натощак и гепатомегалию. Некоторые БНГ (III, IV, IX типы) м.б. связаны с циррозом печени.
Поражаться могут и др. органы, что проявляется в виде нарушения функции почек при I типе, миопатии (скелетной и/или кардиомиопатии) при III и IV типах, а также в некоторых редких формах недостаточности киназы фосфорилазы. Неврологические нарушения возникают при III типе (поражаются периферические нервы) и IV типе (диффузная дисфункция центральной и периферической НС).
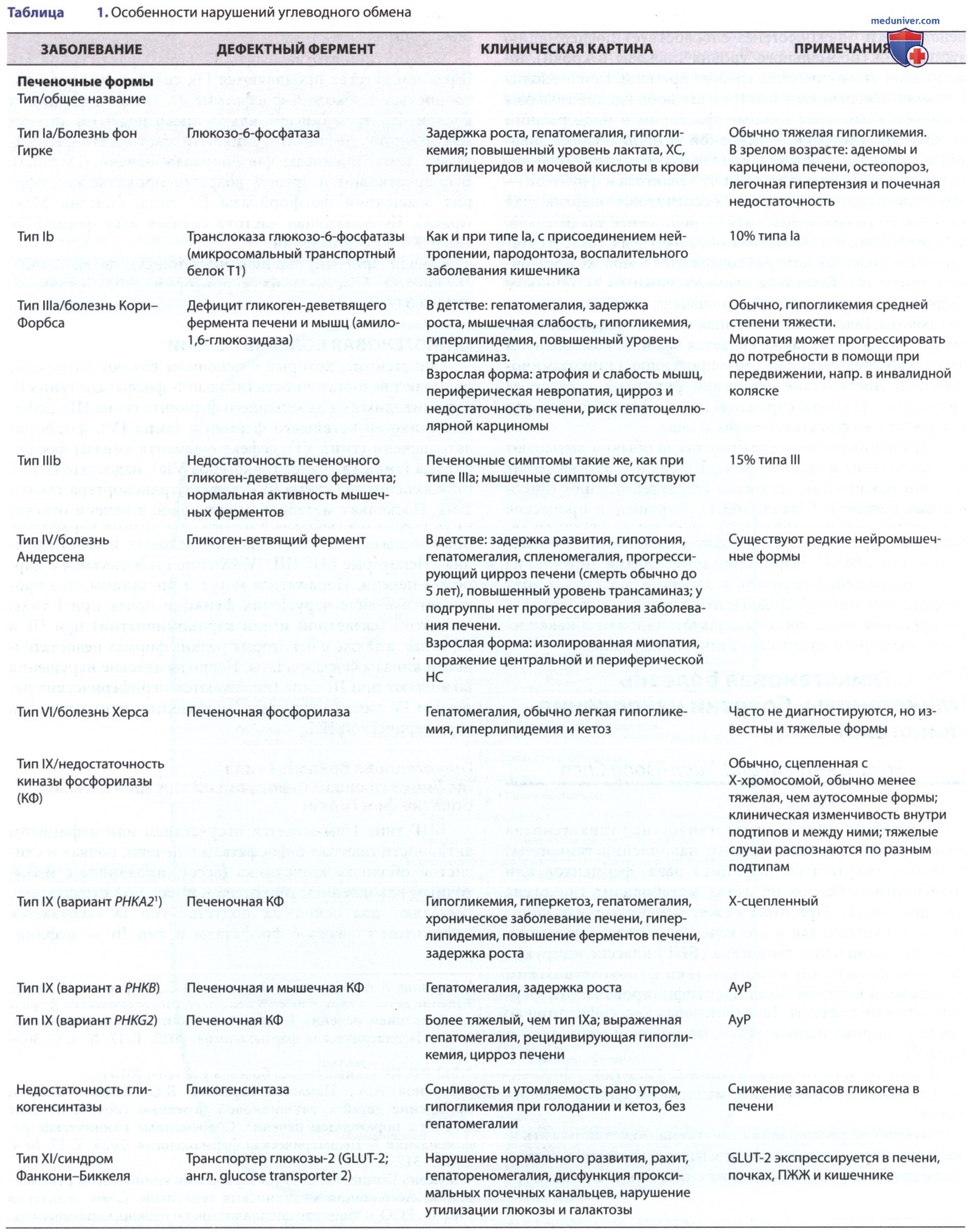
а) Гликогеновая болезнь I типа (дефицит глюкозо-6-фосфатазы или транслоказы, болезнь фон Гирке). БНГ типа I вызывается отсутствием или дефицитом активности глюкозо-6-фосфатазы в печени, почках и слизистой оболочке кишечника (ассоциированное с избыточным накоплением гликогена нормальной структуры). Выделяют два основных подтипа: тип Iа вызывается дефицитом глюкозо-6-фосфатазы и тип Ib — дефицитом микросомального транспортного белка Т1 (транслоказы глюкозо-6-фосфатазы), которая транспортирует глюкозо-6-фосфат через микросомальную мембрану.
Недостаточность ферментов как типа 1а, так и типа 1b приводит к неадекватной печеночной конверсии глюкозо-6-фосфата в глюкозу в результате нормального гликогенолиза и глюконеогенеза, что приводит к гипогликемии при голодании.
БНГ типа I является АуР-заболеванием. Ген глюкозо-6-фосфатазы (G6PC) расположен на хромосоме 17q21; ген транслоказы (SLC37A4) находится на хромосоме Hq23. Выявлены общие патогенные варианты. Анализ ДНК позволяет выявлять носителей генов болезни Гирке и осуществлять ее пренатальную диагностику.
1. Клинические проявления. БНГ типа I могут проявляться гипогликемией и лактатацидозом у новорожденных, но чаще обнаруживается в возрасте 3-4 мес за счет появления гепатомегалии и/или гипогликемических припадков. Для детей характерно кукольное лицо с пухлыми щеками, относительно тонкими конечностями, низкорослость и выпяченный из-за гематомегалии живот. Почки также увеличены, но селезенка и сердце нормальные.
Биохим. характеристиками БНГ типа I являются гипогликемия, лактатацидоз, гиперурикемия и гиперлипидемия. Гипогликемия и лактатацидоз могут развиться после непродолжительного голодания. Гиперурикемия присутствует у маленьких детей; она редко прогрессирует до симптоматической подагры до полового созревания. Несмотря на выраженную гепатомегалию, уровни печеночных трансаминаз обычно нормальны или незначительно повышены. Интермиттирующая диарея может возникать при БНГ типа I.
У пациентов с БНГ типа Ib потеря барьерной функции слизистой оболочки в результате воспаления, которая, вероятно, связана с нарушенной функцией нейтрофилов, по-видимому, является основной причиной диареи. Легкое появление синяков и носовые кровотечения являются обычным явлением и связаны с увеличенным временем кровотечения в результате нарушения агрегации и адгезии тромбоцитов.
Плазма может иметь «молочный» вид из-за резко повышенного уровня триглицеридов. Также повышены ХС и фосфолипиды, но менее выражено. Нарушение липидов напоминает гиперлипидемию типа IV и характеризуется повышенными уровнями ЛПОНП, ЛПНП и уникальным профилем аполипопротеинов, состоящим из повышенных уровней аполипопротеинов В, С и Е с относительно нормальными или пониженными уровнями аполипопротеинов А и D. Гист, вид печени характеризуется увеличенными гепатоцитами за счет переполнения гликогеном и жиром. Липидные вакуоли особенно большие и выраженные. Нет сопутствующего фиброза печени.
Хотя БНГ типа I поражает в основном печень, в патологический процесс вовлекаются и многие др. органы. Часто наблюдается задержка полового созревания. У женщин результаты УЗИ могут соответствовать поликистозу яичников, даже если др. признаки синдрома поликистозных яичников (акне, гирсутизм) отсутствуют. При этом фертильная функция сохраняется, что подтверждено несколькими случаями успешной беременности у женщин с БНГ типа I. Описаны случаи усиления маточных кровотечений во время менструального цикла, включая опасную для жизни меноррагию, что м.б. связано с нарушением агрегации тромбоцитов.
Из-за длительной гиперурикемии в период полового созревания обычно начинаются симптомы подагры. Нарушения липидного обмена повышают риск панкреатитов. Дислипидемия в сочетании с повышенной агрегацией эритроцитов может предрасполагать к развитию атеросклероза. Но случаи раннего атеросклероза описаны крайне редко. Нарушение агрегации тромбоцитов и повышенная антиоксидантная защита для предотвращения перекисного окисления липидов могут действовать как защитный механизм, помогающий снизить риск развития атеросклероза. Часто возникают переломы на фоне рентгенологических признаков остеопении; содержание минералов в костях снижено даже у пациентов препубертатного возраста.
Между второй и третьей декадой жизни у некоторых пациентов с БНГ типа I развиваются аденомы печени, которые могут кровоточить и нередко склонны к озлока-чествлению. У людей, длительно страдающих БНГ I типа, как осложнение может развиться легочная гипертензия. Также часто встречаются железодефицитная анемия и аутоиммунное поражение ЩЖ.
Еще одним поздним осложнением является почечная недостаточность, и у большинства пациентов с БНГ типа I >20 лет наблюдается протеинурия. Многие также страдают АГ, МКБ, нефрокальцинозом и нарушенным клиренсом креатинина. Увеличение СКФ, почечного кровотока и микроальбуминурия часто обнаруживаются на ранних стадиях почечной дисфункции и могут возникать до появления протеинурии. У молодых пациентов увеличение СКФ и почечного кровотока может быть единственным нарушением работы почек. По мере развития почечной недостаточности становятся очевидными фокально-сегментарный гломерулосклероз и интерстициальный фиброз. У некоторых пациентов развивается ХПН, требующая диализа и трансплантации почки.
Др. почечные нарушения включают амилоидоз, синдром Фанкони, гипоцитратурию, гиперкальциурию и дистальноканальцевый ацидоз.
Пациенты с БНГ типа Ib могут иметь дополнительные признаки рецидивирующих бактериальных инфекций, связанных с нейтропенией и нарушением функции нейтрофилов. Поражение полости рта, включая рецидивирующие язвы слизистой оболочки, гингивит и быстро прогрессирующее заболевание пародонта, может возникать при типе Ib. Также при БНГ распространены язвы слизистой оболочки кишечника, достигающие кульминации в виде энтероколита. Тип Ib также связан с хроническим ВЗК, похожим на картину, вовлекающую толстую кишку, которая м.б. связана с нейтропенией и/или дисфункцией нейтрофилов; это может напоминать язвенный колит или болезнь Крона.
2. Диагностика. БНГ типа I можно заподозрить на основании клинических проявлений и лабораторных данных — гипогликемии, лактатацидоза, гиперурикемии и гиперлипидемии. Нейтропения отмечается у пациентов с БНГ типа lb, обычно в возрасте до 1 года. Также нейтропения была отмечена у некоторых пациентов с БНГ типа Ia, особенно у пациентов с вариантом p.G188A. Введение глюкагона или эпинефрина («Адреналина») практически не повышает уровень глюкозы в крови, но концентрация лактата при этом значительно возрастает. Прежде чем стало возможно генетическое тестирование, для окончательного диагноза потребовалась биопсия печени.
Вариантный анализ на основе генов с помощью секвенирования одного гена или генных панелей обеспечивает неинвазивный способ диагностики большинства пациентов с гликогенозом типов Iа и Ib.
В РФ окончательная верификация БНГ проводится с помощью молекулярно-генетического исследования в референсном федеральном центре.
3. Лечение. Лечение направлено на поддержание нормального уровня глюкозы в крови. Это достигается либо введением глюкозы через назогастральный зонд, либо пероральным приемом сырого кукурузного крахмала. В младенчестве для поддержания нормогликемии может потребоваться капельное кормление в течение ночи. Питание через назогастральный зонд может состоять из специальной смеси, только глюкозы или полимера глюкозы, чтобы обеспечить достаточное ее количество для поддержания эутликемии. Обычно в течение дня достаточно частых кормлений с высоким содержанием углеводов.
Сырой кукурузный крахмал действует как форма глюкозы с медленным высвобождением и может вводиться 1,6 г/кг Q4H для детей <2 лет. Наблюдается разл. реакция у маленьких детей. Для детей старшего возраста режим приема кукурузного крахмала увеличивается и составляет 1,6-2,5 г/кг Q4-6H* внутрь в жидком виде. Считается, что новые продукты, содержащие крахмал, такие как восковидный кукурузный крахмал с пролонгированным высвобождением, обладают более длительным действием, лучше переносятся и имеют более приятный вкус. Добавка среднецепочечных триглицеридов цепи улучшает метаболический контроль, что приводит к улучшению роста у детей.
Поскольку фруктоза и галактоза не могут быть напрямую преобразованы в глюкозу при БНГ типа I, эти сахара следует ограничить в рационе. Следует избегать или ограничивать сахарозу (столовый сахар, тростниковый сахар, др. ингредиенты), фруктозу (фрукты, сок, кукурузный сироп с высоким содержанием фруктозы), лактозу (молочные продукты) и сорбитол. В результате этих диетических ограничений может возникнуть дефицит витаминов и минералов, таких как кальций и витамин D, что требует коррекции по витаминам и микроэлементам.
P.S. * МЗ РФ. КР «Гликогеновая болезнь у детей», 2016 г.
Диетотерапия уменьшает степень гиперурикемии, гиперлипидемии и улучшает функцию почек, замедляя развитие почечной недостаточности. Однако эта терапия не может полностью нормализовать уровни мочевой кислоты и липидов в крови у некоторых людей, несмотря на хороший метаболический контроль, особенно после полового созревания. Контроль над гиперурикемией м.б. дополнительно усилен применением аллопуринола, ингибитора ксантиноксидазы. Гиперлипидемию можно снизить с помощью гиполипидемических ЛП, таких как ингибиторы β-гидрокси-β-метилглутарил-кофермента A (HMG-KoA) редуктазы и производные фиброевой кислоты.
Микроальбуминурия, ранний индикатор нарушения почек при болезни типа I, лечится иАПФ. Добавки цитрата могут быть полезны пациентам с гипоцитратурией, предотвращая или уменьшая нефрокальциноз и развитие мочевых камней. Тиазидные диуретики увеличивают реабсорбцию фильтруемого кальция почками и уменьшают выведение кальция с мочой, тем самым предотвращая гиперкальциурию и нефрокальциноз. СТГ следует использовать с особой осторожностью и ограничиваться только теми пациентами, у кого есть документально подтвержденный дефицит СТГ. Даже у этих пациентов следует тщательно контролировать метаболические параметры и наличие аденом.
У пациентов с БНГ типа Ib гранулоцитарные и гранулоцитарно-макрофагальные колониестимулирующие факторы успешно корректируют нейтропению, уменьшая количество и тяжесть бактериальных инфекций и улучшая хроническую ВЗК. Следует использовать минимальную эффективную дозу, поскольку у этих агентов отмечены побочные эффекты, включая спленомегалию, гиперспленизм и боль в костях. Сообщалось, что трансплантация костного мозга корректирует нейтропению БНГ типа Ib.
Ортотопическая трансплантация печени является потенциальным лекарством от БНГ типа I, особенно для пациентов с ЗНО печени, множественными аденомами печени, метаболическими нарушениями, не поддающимися лечению, и печеночной недостаточностью. Однако это следует рассматривать как последнее средство из-за присущих кратко- и долгосрочных осложнений. Большие аденомы (>2 см), которые быстро увеличиваются в размере и/или количестве, могут потребовать частичной резекции печени. Аденомы меньшего размера (<2 см) можно лечить с помощью чрескожной инъекции этанола или транскатетерной артериальной эмболизации. Рецидив аденомы печени является проблемой и может усилить злокачественную трансформацию у этих пациентов, что в конечном счете потребует трансплантации печени.
Перед любой хирургической процедурой необходимо оценить состояние свертывающей системы крови и тщательно компенсировать метаболические нарушения. Длительное кровотечение можно нормализовать с помощью интенсивной в/в инфузии глюкозы за 24-48 ч до операции. 1-деамино-8-D-аргинин вазопрессин может уменьшить кровотечение, но его следует использовать с осторожностью из-за риска гиперволемии и гипонатриемии при в/в введении. Следует избегать приема натрия лактата раствора сложного [Калия хлорида + Кальция хлорида + Натрия хлорида + Натрия лактата] («Рингер лактата»), поскольку он содержит лактат и не содержит глюкозы. Уровень глюкозы должен поддерживаться в пределах нормы на протяжении всей операции с использованием 10% декстрозы («Глюкозы»).
В целом метаболический контроль оценивается по росту, улучшению и коррекции метаболических нарушений, таких как повышенный уровень лактата, глюкозы, триглицеридов, ХС и мочевой кислоты.
4. Прогноз. Ранее БНГ типа I ассоциировался с высокой смертностью в молодом возрасте, и даже для тех, кто выздоровел, прогноз был неопределенный. У больных, получавших в детстве неадекватное лечение, во взрослом возрасте развивались серьезные осложнения. Благодаря ранней диагностике и эффективному лечению существенно улучшились прогнозы заболевания. Однако сохраняются серьезные осложнения, такие как заболевание почек и образование аденом печени с потенциальным риском злокачественной трансформации. Остается серьезной проблемой своевременная идентификация трансформации аденомы печени в гепатоцеллюлярную карциному, так как уровни АФП и карциноэмбрионального АГн часто остаются нормальными в условиях гепатоцеллюлярной карциномы.
б) Гликогеновая болезнь типа III (дефицит гликогендеветвящего фермента, лимитдекстриноз). БНГ типа III (дефицит гликоген-деветвящего фермента «дебранчера», лимитдекстриноз, болезнь Кори, болезнь Форбса, дефицит амило-1,6-глюкозидазы) вызывается недостаточной активностью фермента, расщепляющего гликоген. Гликоген-деветвящий фермент вместе с фосфорилазой отвечает за полное расщепление гликогена. При дефекте деветвящего фермента распад гликогена неполный, что приводит к накоплению аномального гликогена с укороченными боковыми цепями, которые напоминают остаточный декстрин.
Симптомы дефицита деветвящего фермента включают гепатомегалию, гипогликемию, низкорослость, ту или иную степень миопатии и кардиомиопатию. БНГ типа IIIа обычно поражает как печень, так и мышцы, тогда как при типе IIIb, наблюдаемом у 15% пациентов, болезнь поражает только печень.
БНГ типа III является АуР-заболеванием и встречается у разных этнических групп. Частота относительно высока у евреев-сефардов из Северной Африки, жителей Фарерских островов и инуитов. Ген расщепления фермента расположен на хромосоме 1р21. Выявлено более 130 разл. патогенных вариантов; два патогенных варианта в экзоне 3, c.18_19delGA (ранее описанный как c.17_18delAG) и p.Gln6X специфически связаны с БНГ типа IIIb. Анализ ДНК и выявление специфических мутаций позволяют обнаружить носителей гена и осуществлять пренатальную диагностику.
1. Клинические проявления. В периоде новорожденности и детстве БНГ типа III м.б. неотличим от БНГ типа I, в обоих случаях наблюдаются гепатомегалия, гипогликемия, гиперлипидемия и задержка роста (рис. 2). Может наблюдаться спленомегалия, но почки обычно не поражаются. Гепатомегалия у большинства пациентов с БНГ типа III улучшается с возрастом; однако фиброз печени, цирроз, прогрессирующий до печеночной недостаточности, и гепатоцеллюлярная карцинома отмечаются у многих в позднем возрасте. Аденомы печени встречаются реже у людей с БНГ типа III, чем у людей с БНГ типа I. Связь между аденомами печени и ЗНО при БНГ типа III остается неясной.
Рисунок 2. Рост и развитие пациента с болезнью накопления гликогена типа IIIb. У пациента дефицит деветвящего фермента в печени, но нормальная активность в мышцах. В детстве отмечалась гепатомегалия, гипогликемия и задержка роста. После полового созревания гепатомегалия или гипогликемия исчезли; окончательный рост нормальный. Слабость и атрофия мышц отсутствуют. Это отличается от клинической картины болезни накопления гликогена с типом IIIa, при котором наблюдается прогрессирующая миопатия во взрослом возрасте
Уровни АФП и карциноэмбрионального АГн не являются надежными предикторами наличия гепатоцеллюлярных аденом или злокачественной трансформации. Отмечен единичный случай злокачественной трансформации на месте аденом.
Мышечная слабость у пациентов с БНГ типа IIIа медленно прогрессирует и связана с истощением. Мышечная слабость практически незаметна в детстве, но может стать очевидной после 3-4-й декады жизни. Низкая минеральная плотность костной ткани у пациентов с БНГ типа III подвергает их повышенному риску потенциальных переломов. Миопатия не следует какой-либо конкретной схеме поражения; задействованы как проксимальные, так и дистальные мышцы. ЭМГ выявляет распространенную миопатию; нарушается проведение по нервам.
Хотя явная сердечная дисфункция возникает редко, часто встречается гипертрофия желудочков. При сердечной патологии выявлено диффузное поражение разл. структур сердца, включая вакуолизацию миоцитов, нарушение АВ-проводимости и гиперплазию гладких мышц. У некоторых пациентов с БНГ типа III сообщалось об опасной для жизни аритмии и необходимости трансплантации сердца. Печеночные симптомы у некоторых пациентов м.б. настолько легкими, что диагноз ставится только в зрелом возрасте, когда у пациентов появляются симптомы и признаки нервно-мышечных нарушений.
Начальные проявления можно принять за болезнь Шарко-Мари-Тута. Отмечаются поликистоз яичников; у некоторых пациенток может развиться гирсутизм, нерегулярный менструальный цикл и др. признаки СПКЯ. На фертильность не влияет; сообщалось об успешных беременностях.
Часто встречаются гипогликемия и гиперлипидемия. В отличие от БНГ типа Iа, отмечается значительное повышение уровня печеночных трансаминаз и кетоновых тел натощак, но концентрации лактата и мочевой кислоты в крови обычно в норме. Введение глюкагона через 2 ч после приема углеводной еды вызывает нормальное повышение уровня глюкозы в крови; однако после ночного голодания глюкагон может не вызывать изменения уровня глюкозы в крови. О поражении мышц судят по повышению КФК в сыворотке крови, хотя нормальные ее уровни не исключают дефицит деветвящего фермента в мышцах.
2. Диагностика. При гист. исследовании печени характерны переполненные гликогеном гепатоциты и фиброзные перегородки. Фиброз и недостаток жира отличают БНГ типа III от типа I. Фиброз м.б. минимальным и захватывать лишь перипортальную область или достигать микронодулярного цирроза, но в большинстве случаев, по-видимому, не прогрессирует. Открытый цирроз печени наблюдался у некоторых пациентов с БНГ типа III.
Пациенты с миопатией и печеночными симптомами имеют генерализованный ферментный дефект (тип IIIa). Недостаточная активность фермента наблюдается не только в печени и мышцах, но и в др. тканях, таких как сердце, эритроциты и в культуре фибробластов. Пациенты с печеночными симптомами без клинических или лабораторных признаков миопатии имеют дефицит расщепляющих ферментов только в печени, при этом активность ферментов в мышцах сохранена (тип IIIb). До того, как стали доступны генетические тесты, для окончательного диагноза требовался анализ ферментов в печени, мышцах или и тех и других. Секвенирование генов теперь позволяет диагностировать и определять подтипы у большинства пациентов.
3. Лечение. Основным методом лечения БНГ типа III является диетотерапия, как и при БНГ типа I, хотя она имеет меньшее значение. Пациентам не нужно ограничивать потребление фруктозы и галактозы с пищей, хотя следует избегать простых сахаров, чтобы предотвратить внезапные скачки уровня глюкозы в крови. Гипогликемию лечат небольшими частыми приемами пищи с высоким содержанием сложных углеводов, напр. добавками кукурузного крахмала или ночным назогастральным капельным кормлением. Кроме того, для предупреждения развития гипогликемии в дневное время эффективна диета с высоким содержанием белка, а также энтеральная инфузия белка на ночь. Экзогенный белок можно использовать в качестве субстрата для глюконеогенеза, который помогает удовлетворить потребности в энергии и предотвратить распад эндогенного белка. Белок в рационе также снижает общую потребность в крахмале.
Следует избегать чрезмерного приема кукурузного крахмала, поскольку это может привести к чрезмерному накоплению гликогена, что неблагоприятно и может привести к чрезмерному увеличению МТ. Добавка среднецепочечных триглицеридов цепи рассматривается как альтернативный источник энергии. Не существует успешного лечения прогрессирующей миопатии, кроме рекомендации диеты с высоким содержанием белка и программы субмаксимальных нагрузок. Для выявления прогрессирования фиброза печени в цирроз и далее в гепатоцеллюлярную карциному необходим тщательный мониторинг МРТ БП. Трансплантация печени выполнялась пациентам с БНГ типа III с прогрессирующим циррозом и/или гепатоцеллюлярной карциномой. Имеются сообщения о трансплантации сердца у пациентов с БНГ типа III с терминальной стадией заболеваний сердца.
в) Гликогеновая болезнь типа IV. БНГ типа IV (болезнь Андерсен, дефицит амило-1,4:1,6-глюкантрансферазы, дефицит гликоген-ветвящего фермента, амилопектиноз) вызывается недостаточностью активности фермента ветвления, что приводит к накоплению аномального плохо растворимого гликогена. Заболевание также известно как амилопектиноз, потому что аномальный гликоген имеет меньше точек ветвления, больше а-1,4-гликозопептидных связей и более длинные внешние цепи, что приводит к образованию аномальной структуры, напоминающей амилопектин. Накопление полиглюкозана, положительного при взаимодействии с реактивом Шиффа и частично устойчивого к амилазе, наблюдается во всех тканях пациентов, но в разной степени.
БНГ типа IV является АуР-заболеванием. Ген гликоген-ветвящего фермента (GBE; англ. glycogen-branching enzyme) расположен на хромосоме 3p21. Было идентифицировано >20 патогенных вариантов, ответственных за БНГ типа IV, а их характеристика в каждом отдельном случае позволяет предвидеть клиническую картину заболевания у пациента. Почти полное отсутствие активности GBE с нулевыми исходами было связано с перинатальной смертью и фатальной неонатальной гипотонией. Остаточная активность GBE >5% и наличие по крайней мере одного миссенс-варианта связаны с фенотипом нелетального цирроза печени и, в некоторых случаях, с отсутствием прогрессирующего заболевания печени.
1. Клинические проявления. Клиническая картина при БНГ типа IV очень вариабельна. Наиболее распространенная и классическая форма характеризуется прогрессирующим циррозом печени и проявляется в первые 18 мес жизни как гепатоспленомегалия и нарушение нормального развития. Цирроз может проявляться портальной гипертензией, асцитом и варикозным расширением вен пищевода и может прогрессировать до печеночной недостаточности, обычно приводящей к смерти к 5-летнему возрасту. В редких случаях заболевания печени не прогрессируют; у таких пациентов отмечается умеренное изменение структуры печени, и они не требуют пересадки печени.
Внепеченочное поражение у некоторых пациентов с БНГ типа IV включает поражение опорно-двигательного аппарата, в частности вовлечение сердечных мышц и мышц скелета, а также поражение ЦНС.
Сообщалось о нервно-мышечной форме БНГ типа IV с четырьмя основными вариантами, распознаваемыми в зависимости от возраста на момент обращения. Перинатальная форма характеризуется последовательностью акинезии и деформации плода и смертью в перинатальном периоде. Врожденная форма проявляется при рождении с тяжелой гипотонией, мышечной атрофией и поражением нейронов, смертью в неонатальном периоде; у некоторых пациентов присутствует кардиомиопатия. Детская форма проявляется в основном миопатией или кардиомиопатией. Взрослая форма, болезнь полиглюкозановых телец у взрослых, проявляется в виде изолированной миопатии или диффузного поражения ЦНС и периферической НС, сопровождающейся накоплением полиглюкозановых телец в нейронах.
Симптомы поражения нейронов включают периферическую невропатию, нейрогенный мочевой пузырь и лейкодистрофию, а также умеренное снижение когнитивных функций у некоторых пациентов.
При болезни полиглюкозановых телец у взрослых для установления диагноза необходимо определение уровня гликоген-ветвящего фермента в лейкоцитах или биоптатах нервной ткани, поскольку его недостаточность ограничена именно этими клетками.
2. Диагностика. При БНГ типа IV отложение атипичного амилопектин-подобного гликогена выявляется в печени, сердце, мышцах, коже, кишечнике, ГМ, спинном мозге и периферических нервах. В печени развивается мелкоузловой цирроз. При гист. исследовании гепатоцитов видны слабо окрашенные базофильные включения в гепатоцитах, которые представляют собой крупнозернистые ШИК*-позитивные отложения, частично устойчивые к амилазе. Электронная микроскопия показывает, помимо обычных частиц гликогена α и β, накопление волокнистых агрегатов, типичных для амилопектина. Характерное окрашивание цитоплазматических включений, а также результаты электронной микроскопии могут иметь диагностическое значение, однако аналогичные гист. признаки наблюдались при полисахаридозах, не сопровождающихся недостаточностью гликоген-ветвящего фермента.
Для окончательной постановки диагноза необходимо определить содержание этого фермента в печени, мышцах, культуре фибробластов кожи или лейкоцитах или идентифицировать патогенный вариант в гене GBE. Пренатальная диагностика возможна путем измерения активности ферментов в культивируемых амниоцитах, ворсинах хориона или с помощью методик на основе ДНК.
P.S. * ШИК (Periodic acid-Schiff, PAS) — метод окрашивания Шифф-йодной кислотой, используемый в гистологии и патологии для выявления гликогена в тканях.
3. Лечение. Специального лечения БНГ типа IV не существует. Поражение НС, напр. проблемы с походкой и поражение мочевого пузыря, требует симптоматического лечения. В отличие от пациентов с др. БНГ печени (типы I, III, VI, IX), пациенты с БНГ типа IV не имеют гипогликемии, которая наблюдается только при явном циррозе печени. Трансплантация печени выполнялась пациентам с прогрессирующим заболеванием печени, но пациенты должны быть тщательно отобраны, поскольку это муль-тисистемное заболевание и у некоторых пациентов внепеченочные нарушения могут проявиться после трансплантации. Неизвестны случаи долгосрочного успеха трансплантации печени. У лиц со значительным диффузным вовлечением ретикулоэндотелия высокий риск заболеваемости и смертности, что может повлиять на эффективность трансплантации печени.
г) Гликогеновая болезнь типа VI. БНГ VI типа [болезнь Херса (Эра), дефицит фосфорилазы печени] вызывается дефицитом печеночной фосфорилазы. В настоящий момент зарегистрировано небольшое количество пациентов, что, вероятно, связано с занижением данных по этому заболеванию. Болезнь проявляется в раннем детстве гепатомегалией и задержкой роста. Гипогликемия, гиперлипидемия и кетоацидоз имеют разную степень выраженности. Кетотическая гипогликемия может проявиться после ночного или длительного голодания. Уровни молочной и мочевой кислот в норме. БНГ VI типа имеет широкий спектр проявлений, у некоторых пациентов м.б. выраженные клинические проявления.
Сообщалось о пациентах с тяжелой гепатомегалией, рецидивирующей тяжелой гипогликемией, кетоацидозом и постпрандиальным лактатацидозом. У некоторых больных отмечается очаговая узловая гиперплазия печени и гепатоцеллюлярная аденома с трансформацией в карциному.
Ранее считалось, что отсутствует поражение миокарда, но недавно описан случай выявления легкой кардиомиопатии у пациента с БНГ типа VI.
Лечение симптоматическое и направлено на предотвращение гипогликемии при обеспечении адекватного питания. Диета с высоким содержанием углеводов и белков и частое кормление эффективно предотвращают гипогликемию. Следует регулярно контролировать уровень глюкозы и кетонов в крови, особенно в периоды повышенной активности/болезни. Требуется долгосрочное наблюдение за этими пациентами для понимания течения заболевания.
БНГ типа VI является АуР-заболеванием. Диагноз можно подтвердить с помощью молекулярного тестирования гена фосфорилазы печени (PYGL; англ. Glycogen phosphorylase), который обнаружен на хромосоме 14q21-22 и имеет 20 экзонов. Известно много патогенных вариантов этого гена; вариант сайта сращивания в интроне 13 был идентифицирован в популяции меннонитов. Также для постановки диагноза может использоваться биопсия печени, показывающая повышенное содержание гликогена и пониженную активность печеночной фосфорилазы. Однако, благодаря возможности анализа ДНК и панелям секвенирования следующего поколения, биопсия печени считается ненужной.
д) Гликогеновая болезнь типа IX (недостаточность киназы фосфорилазы). БНГ типа IX представляет собой гетерогенную группу гликогенозов. Это происходит из-за недостаточности фермента киназы фосфорилазы (КФ), который участвует в лимитирующей стадии гликогенолиза. Этот фермент имеет четыре субъединицы (α, β, γ, δ), каждая из которых кодируется разными генами на разных хромосомах и по-разному экспрессируется в разных тканях. Патогенные варианты гена РНКА1 вызывают недостаточность КФ в мышцах; патогенные варианты генов РНКА2 и PHKG2 — недостаточность КФ в печени; патогенные варианты гена РНКВ — недостаточность КФ в печени и мышцах. Патогенные варианты в гене PHKG1 не идентифицированы. Дефекты в субъединицах α, β и γ ответственны за реакции печени.
Клинические проявления недостаточности КФ печени обычно распознаются в течение первых двух лет жизни и включают задержку роста, вздутие живота, связанное с умеренной или выраженной гепатомегалией. Клиническая тяжесть значительно различается и зависит от недостаточности КФ в печени. Гиперкетотическая гипогликемия, если присутствует, м.б. легкой, но в редких случаях м.б. тяжелой. Кетоз может возникнуть даже при нормальном уровне глюкозы. У некоторых детей м.б. небольшая задержка физического развития и мышечная гипотония. Становится все более очевидным, что БНГ типа IX не является доброкачественным заболеванием. Сообщается о тяжелых фенотипах с фиброзом печени, прогрессирующим до цирроза и гепатоцеллюлярной карциномы, особенно у пациентов с вариантами PHKG2. Сообщается о прогрессирующей спленомегалии и портальной гипертензии как осложнениях цирроза печени.
Описан случай легкой кардиомиопатии у пациента с БНГ типа IX (вариант РНКВ). Когнитивные и речевые задержки были зарегистрированы у нескольких человек, но неясно, вызваны ли эти задержки недостаточностью КФ или они случайны. В редких случаях сообщалось о почечном канальцевом ацидозе. В отличие от БНГ типа I, лактатацидоз, склонность к кровотечениям и жидкий стул не характерны. Несмотря на то что в детстве рост замедляется, в конечном счете достигается нормальный рост и полное половое развитие. Как и в случае дефицита гликоген-деветвящего фермента, увеличение живота и гепатомегалия обычно проходят с возрастом и могут исчезнуть к подростковому возрасту. У большинства взрослых с недостаточностью КФ печени симптомы отсутствуют, хотя необходимы дальнейшие долгосрочные исследования, чтобы полностью оценить влияние этого расстройства на взрослых.
Фенотипическая изменчивость внутри каждого подтипа раскрывается с помощью молекулярного тестирования.
1. Сцепленная с Х-хромосомой недостаточность киназы фосфорилазы печени (из вариантов РНКА2). Сцепленная с Х-хромосомой недостаточность КФ печени является одной из наиболее частых форм гликогеноза печени у мужчин. Помимо печени, активность фермента также м.б. снижена в эритроцитах, лейкоцитах и фибробластах при нормальном содержании в мышцах. В типичных случаях у мальчика 1-5 лет наблюдаются задержка роста, гепатомегалия как случайная находка и небольшая задержка развития моторики. Незначительно повышены ХС, триглицериды и печеночные ферменты. Кетоз может возникнуть после голодания. Уровни лактата и мочевой кислоты в норме. Гипогликемия обычно если присутствует, то в легкой форме, но м.б. и тяжелой.
Реакция глюкозы в крови на глюкагон нормальная. Гепатомегалия и изменения в биохим. показателях крови постепенно улучшаются и могут нормализоваться с возрастом. Большинство взрослых достигают нормального конечного роста и обычно не имеют симптомов, несмотря на стойкую недостаточность КФ.
Все чаще признается, что это заболевание не является доброкачественным, как считалось ранее, и есть пациенты с тяжелым течением заболевания и долгосрочными последствиями для печени. В редких случаях может развиться фиброз печени, переходящий в цирроз.
При гист. исследовании печени обнаруживаются перегруженные гликогеном гепатоциты, стеатоз и, возможно, умеренный перипортальный фиброз. Агрегаты гликогена (β-частицы в форме розеток) имеют более рыхлый или лопнувший вид и менее компактны, чем гликоген, наблюдаемый при БНГ типа I или III. Можно увидеть образование фиброзной перегородки и незначительные воспалительные изменения.
Ген общей печеночной изоформы α-субъединицы КФ, РНКА2, расположен на Х-хромосоме (aL в Хр22.2). Мутации в гене РНКА2 составляют 75% всех случаев КФ. Сцепленная с Х-хромосомой недостаточность КФ печени далее подразделяется на два биохимических подтипа: XLG1, с измеримым дефицитом активности КФ как в клетках крови, так и в печени, и XLG2, с нормальной активностью КФ in vitro в клетках крови и переменной активностью в печени. Предполагается, что XLG2 м.б. вызван миссенс-вариантами, которые влияют на регуляцию фермента, тогда как нонсенс-варианты, влияющие на количество белка, приводят к XLG1. Женщины-носители не страдают от этого заболевания.
2. Аутосомная недостаточность печеночной и мышечной киназы фосфорилазы (из вариантов РНКВ) [гликогеновая болезнь IXb типа (синонимы: дефицит мышечной/печеночной киназы фосфорилазы]. Описаны случаи недостаточности КФ в клетках печени и крови с АуР-типом наследования. Подобно сцепленной с Х-хромосомой форме, главными симптомами в раннем детстве являются гепатомегалия и задержка роста. У некоторых пациентов также наблюдается мышечная гипотония. В нескольких случаях, когда была измерена активность фермента, в мышцах наблюдалось снижение активности КФ. Мутации обнаруживаются в РНКВ (хромосома 16q12-q13), кодирующем субъединицу β, и приводят к дефициту КФ в печени и мышцах. Были идентифицированы несколько нонсенс-вариантов, вставка одного основания, мутация сайта сращивания и большая внутригенная мутация. Кроме того, у нетипичного пациента с нормальной активностью КФ клеток крови был обнаружен миссенс-вариант.
3. Аутосомная недостаточность печеночной киназы фосфорилазы (из вариантов PHKG2) (гликогеновая болезнь IXc-типа, недостаточность тестикулярной/ печеночной изоформы γ-субъединицы киназы фосфорилазы). Эта форма недостаточности КФ вызвана патогенными вариантами тестикулярной/печеночной изоформы гена субъединицы у (PHKG2). В отличие от сцепленной с Х-хромосомой недостаточности ФК, пациенты с вариантами PHKG2 обычно имеют более тяжелые фенотипы с рецидивирующей гипогликемией, выраженной гепатомегалией, значительным фиброзом печени и прогрессирующим циррозом. Поражение печени может проявляться холестазом, пролиферацией желчных протоков, варикозным расширением вен пищевода и спленомегалией. Кроме того, описаны клинические симптомы, включающие задержку моторного развития, мышечную гипотонию и повреждение почечных канальцев.
Спектр проявлений продолжает расширяться по мере увеличения количества наблюдаемых пациентов. PHKG2 расположен на хромосоме 16р12.1-р11.2; известно множество патогенных вариантов этого гена.
4. Недостаточность киназы фосфорилазы миокарда. У этих пациентов уже в грудном возрасте наблюдалась кардиомиопатия, которая быстро прогрессировала и приводила к смерти от СН. Недавние исследования показали, что это не случай специфической для сердца первичной недостаточности КФ, как предполагалось ранее; он скорее связан с субъединицей γ2-аденозинмонофосфат (АМФ)-активированной протеинкиназы (см. ниже). Субъединица γ2 кодируется геном PRKAG2.
5. Диагностика. Недостаточность КФ м.б. диагностирована путем выявления ферментативного дефекта в пораженных тканях. КФ можно измерить в лейкоцитах и эритроцитах, но, поскольку фермент имеет много изоферментов, диагноз можно легко пропустить, если не исследовать печень, мышцы или сердце. Лица с недостаточностью КФ печени также обычно имеют повышенные трансаминазы, слегка повышенные триглицериды и ХС, нормальные концентрации мочевой и молочной кислоты, а также нормальный ответ глюкагона. Секвенирование генов используется для диагностического подтверждения и определения подтипов БНГ типа IX.
Наиболее часто участвует ген РНКА2, кодирующий субъединицу а, за которым следует ген РНКВ, кодирующий субъединицу β. Варианты гена PHKG2, лежащие в основе дефицита γ-субъединицы, обычно связаны с тяжелым поражением печени с рецидивирующей гипогликемией и фиброзом печени.
6. Лечение и прогноз. Лечение недостаточности КФ печени симптоматическое. Оно включает диету с высоким содержанием сложных углеводов и белков, а также частые кормления небольшими порциями для предотвращения гипогликемии. Кукурузный крахмал можно вводить в зависимости от симптомов и времени (0,6-2,5 г/кг Q6H). Прием глюкозы внутрь, если он переносится, следует использовать для лечения гипогликемии. Если нет, следует ввести глюкозу в/в.
Прогноз для сцепленных с Х-хромосомой и некоторых аутосомных форм обычно хороший; однако могут быть выявлены отсроченные осложнения. Пациенты с мутациями в субъединице у обычно имеют более тяжелое клиническое течение с прогрессирующим заболеванием печени. У всех пациентов с БНГ типа IX необходимо контролировать вовлечение печени с помощью периодической визуализации (УЗИ БП или МРТ каждые 6-12 мес) и серийных функциональных тестов печени.
е) Недостаточность гликогенсинтазы в печени. Недостаточность гликогенсинтазы печени типа 0 (гликогеновая болезнь типа 0) вызван недостаточностью активности печеночной гликогенсинтазы (GYS2), что приводит к заметному снижению запасов гликогена в печени. Ген GYS2 расположен на 12р12.2. У пациентов с БНГ 0 типа было идентифицировано несколько патогенных вариантов. У людей заболевание встречается редко, и в истинном смысле это не тип гликогеновой болезни, поскольку недостаточность фермента приводит к уменьшению запасов гликогена. Маленькие дети утром (до завтрака) выглядят сонливыми и бледными, у них развивается рвота и быстрая утомляемость, а иногда и судороги, связанные с гипогликемией и кетонемией. Содержание лактата и аланина в крови низкое, гиперлипидемии или гепатомегалии нет.
Длительная гипергликемия, глюкозурия, лактатацидоз и гипераланинемия при нормальном уровне инсулина после приема глюкозы или еды указывают на дефицит гликогенсинтазы.
Окончательный диагноз м.б. поставлен на основе биопсии печени для измерения активности фермента или идентификации патогенных вариантов GYS2.
Лечение состоит из частых приемов пищи, богатых белком, и ночного приема сырого кукурузного крахмала для предотвращения гипогликемии и гиперкетонемии. Большинство детей с БНГ типа 0 отличаются нормальными когнитивными способностями и развитием. К общим чертам можно отнести низкий рост и остеопению. Прогноз благоприятный для пациентов, доживших до взрослого возраста, включая разрешение гипогликемии, за исключением периода беременности.
ж) Гликогеновая болезнь печени с почечным синдромом Фанкони (болезнь накопления гликогена типа XI, синдром Фанкони-Бикеля). Синдром Фанкони-Бикеля является редким АуР-заболеванием, вызванным дефектами в транспортере глюкозы 2 (GLUT-2), который транспортирует глюкозу в и из гепатоцитов, β-клеток ПЖЖ и базолатеральных мембран эпителиальных клеток кишечника и почек. Заболевание характеризуется дисфункцией проксимальных почечных канальцев, нарушением утилизации глюкозы и галактозы и накоплением гликогена в печени и почках.
На первом году жизни у больных детей имеются задержка физического развития, признаки рахита, выпяченный живот из-за гепатомегалии и нефромегалии. Заболевание можно спутать с БНГ типа I, поскольку у этих пациентов может развиваться синдром Фанкони. Взрослые обычно имеют низкий рост, карликовость и избыточный жир в области живота и плеч. Пациенты более подвержены переломам из-за ранней генерализованной остеопении. Кроме того, может возникнуть кишечная мальабсорбция и диарея.
Лабораторные данные включают глюкозурию, фосфатурию, генерализованную аминоацидурию, потерю бикарбоната, гипофосфатемию, повышенные уровни ЩФ в сыворотке и рентгенологические признаки рахита. Может присутствовать легкая гипогликемия натощак и гиперлипидемия. Уровни печеночных трансаминаз, молочной и мочевой кислоты в плазме обычно в норме. Толерантность к глюкозе и галактоза нарушена, что объясняется дефектом GLUT-2, препятствующим усвоению этих сахаров печенью. Результаты биопсии ткани показывают заметное накопление гликогена в гепатоцитах и клетках проксимальных почечных канальцев, предположительно из-за измененного транспорта глюкозы из этих клеток. Сообщалось о диффузном гломерулярном мезангиальном расширении наряду с повышенной клубочковой фильтрацией и микроальбуминурией, аналогичными нефропатии при БНГ типа Iа и СД.
Такое состояние встречается редко, причем у 70% пациентов с синдромом Фанкони-Бикеля родители единокровные (близкородственные браки). Большинство пациентов имеют гомозиготные патогенные варианты; некоторые пациенты являются сложными гетерозиготами. Большинство обнаруженных к настоящему времени вариантов предсказывают преждевременное прекращение трансляции. Результирующая потеря С-концевой области белка GLUT-2 предсказывает нефункционирующий транспортер глюкозы с обращенным внутрь сайтом связывания субстрата.
Специфического лечения не существует. Симптоматическое лечение фосфатом и бикарбонатом может привести к улучшению роста. Рост также может улучшиться с помощью компенсации потерь воды, электролитов и витамина D; ограничения приема галактозы; а также диеты, аналогичной диете, используемой при сахарном диабете, с небольшими частыми приемами пищи и адекватным потреблением калорий.