а) Этиология. Дефицит 21-гидроксилазы — причина >90% случаев врожденной дисфункции коры надпочечников. Этот фермент семейства Р450 (CYP21, Р450с21) гидроксилирует прогестерон и 17-гидроксипрогестерон для получения 11-дезоксикортикостерона и 11-дезоксикортизола соответственно (см. рис. ниже). Эти превращения необходимы для синтеза альдостерона и кортизола соответственно.
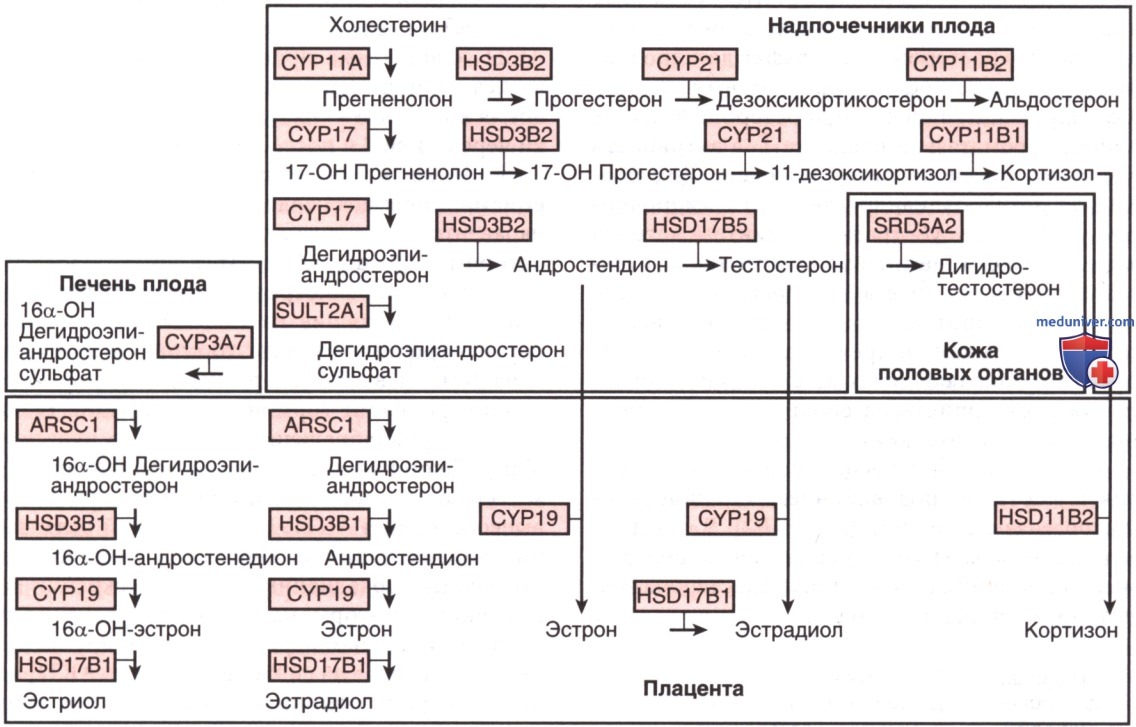
Биосинтез и метаболизм стероидов во время гестации. Превращения в пределах коры надпочечников плода, печени плода, кожи мужских (т.е. подверженных воздействию тестостерона) половых органов и плаценты обозначены стрелками; также показан фермент, опосредующий каждое превращение. Ферментативные превращения в коре надпочечников одинаковы в постнатальный и перинатальный период, однако биосинтез кортизола и альдостерона более выражен, также в норме синтезируется небольшое количество тестостерона. Большинство задействованных ферментов являются представителями семейства цитохрома Р450 (CYP). Ферменты надпочечников включают CYP11A1, фермент, расщепляющий боковую цепь холестерина (прежнее название P450scc); HSD3B2, 3β-гидроксистероид-дегидрогеназу/Δ5,Δ4-изомеразу тип 2; CYP 17, 17β-гидроксилазу/17,20-лиазу (Р450с17); CYP21, 21-гидроксилазу (Р450с21); CYP11B1, 11β-гидроксилазу (Р450с11); CYP11B2, альдостеронсинтетазу (P450aldo; данный фермент опосредует последующие реакции по превращению дезоксикортикостерона в альдостерон с участием 11β-гидроксилазы, 18-гидроксилазы и 18-оксидазы в клубочковой зоне). Другими ферментами, имеющими значение для фетоплацентарной единицы, являются ARSC1, арилсульфатаза; CYP19, ароматаза (P450arom); HSD3B1, Зβ-гидроксистероид-дегидрогеназа/Δ5, Δ4 изомераза тип 1; HSD11В2, 11β-гидроксистероид-дегидрогеназа тип 2; HSD17В1 и HSD17В5 — два различных фермента 17-гидроксистероид-дегидрогеназы; SRD5A2, стероидная 5α-редуктаза тип 2; SULT2A1, стероидная сульфотрансфераза
При наиболее тяжелой, сольтеряющей форме заболевания отмечается дефицит обоих гормонов. У пациентов с немного менее тяжелой формой болезни сохраняется способность синтезировать достаточное количество альдостерона, однако отмечаются повышенные уровни андрогенов надпочечникового происхождения, что обозначается как простая вирильная форма. Эти две формы вместе носят название классического дефицита 21-гидроксилазы. У пациентов с неклассической формой заболевания отмечаются относительно незначительно повышенные уровни андрогенов, бессимптомное течение, могут наблюдаться признаки избытка андрогенов в любом возрасте после рождения. Клиническая картина зависит от генотипа (см. ниже раздел «Генетика») (табл. 2).
б) Эпидемиология. Классический дефицит 21-гидроксилазы возникает в 1:15 000-20 000 рождений в большинстве популяций. Примерно 70% младенцев, страдающих этим заболеванием, имеют сольтеряющую форму, 30% — простую вирильную форму. В США заболевание менее распространено среди афроамериканцев по сравнению с детьми белого населения (1:42 000 и 1:15 500 соответственно). Распространенность неклассической формы заболевания 1:1000 в общей популяции, чаще возникает у представителей отдельных этнических групп, например евреев ашкенази/латиноамериканцев.
в) Генетика. Существует два гена стероид-21-гидроксилазы — CYP21P (CYP21A1P, CYP21A), CYP21 (CYP21A2, CYP21B), которые чередуются в тандеме с двумя генами 4-го компонента комплемента (С4А, С4В) в лейкоцитарном АГн человека (HLA) главного комплекса гистосовместимости на хромосоме 6р21.3 между локусами HLA-B и HLA-DR. В этом кластере располагается множество др. генов. CYP21 — активный ген; CYP21P по последовательности ДНК на 98% идентичен CYP21, но является псевдогеном в связи с девятью различными мутациями. Зарегистрировано почти 300 мутаций, >90% мутантных аллелей, вызывающих дефицит 21-гидроксилазы, что являются результатом рекомбинации между CYP21 и CYP21P. Примерно 20% — делеции, вызванные неравномерным мейотическим кроссинговером между CYP21 и CYP21P, остальные — нереципрокные передачи вредных мутаций от CYP21P к CYP21. Данное явление называется конверсией генов.
Вредные мутации CYP21P оказывают различное влияние на ферментативную активность при переносе на CYP21. Некоторые мутации способствуют полному прекращению синтеза функционального белка, другие — миссенс-мутации (приводят к аминокислотным заменам), в результате которых образуются ферменты с активностью 1-50% от нормальной. Тяжесть заболевания коррелирует с мутациями, носителями которых являются лица с этим заболеванием; например, пациенты с сольтеряющей формой болезни являются носителями мутаций на обоих аллелях, что полностью разрушает ферментативную активность. Пациенты часто гетерозиготные по различным типам мутаций (т.е. один аллель менее сильно поражен, чем другой), в этом случае тяжесть проявления заболевания в значительной степени определяется активностью того из двух аллелей, который наименее затронут.
Прилегая близко к CYP21, но на противоположной цепи ДНК, находится ген tenascin-X (TNX), который кодирует белок соединительной ткани. В редких случаях делеции CYP21 распространяются на TNX. У таких пациентов может наблюдаться синдром «генных последовательностей», состоящий из врожденной гиперплазии надпочечников и синдрома Элерса-Данло.
г) Патогенез и клинические проявления:
1. Дефицит альдостерона и кортизола. Для синтеза кортизола и альдостерона требуется 21-гидроксилирование, при наиболее тяжелой сольтеряющей форме отмечается дефицит обоих этих гормонов. Эта форма составляет 70% всех случаев классического дефицита 21-гидроксилазы. Признаки и симптомы дефицита кортизола и альдостерона и лежащая в их основе патофизиология описаны в главе 593. Они включают прогрессирующую потерю веса, анорексию, рвоту, обезвоживание, слабость, гипотензию, гипогликемию, гипонатриемию, гиперкалиемию. Эти нарушения впервые развиваются у младенцев, страдающих этим расстройством, в возрасте 10-14 дней. Без лечения в течение нескольких дней/недель может возникнуть шок, сердечные аритмии, смерть.
Врожденная дисфункция коры надпочечников отличается от др. причин первичной недостаточности надпочечников тем, что предшественники стероидов накапливаются проксимальнее заблокированного ферментативного превращения. В связи с неэффективным синтезом кортизола возникают высокие уровни АКТГ, что приводит к гиперплазии коры надпочечников, при этом уровни предшественников стероидов могут быть в 100 раз выше нормальных.
При дефиците 21-гидроксилазы эти предшественники включают 17-гидроксипрогестерон и прогестерон. Прогестерон, др. метаболиты действуют как антагонисты минералокортикоидных рецепторов, усугубляя эффекты дефицита альдостерона у пациентов, не получающих лечение.
Нередко дети с классической формой болезни госпитализируются по поводу интеркуррентных заболеваний в детском возрасте. Такая ситуация с наибольшей вероятностью может происходить в течение первых 2 лет жизни и быть вызвана гастроэнтеритом, поскольку такие заболевания могут привести к потере жидкости и электролитов, а рвота может помешать дозированию ЛП. Чаще госпитализируются дети, которым требуются высокие дозы флудрокортизона, т.к. они наиболее предрасположены к потере соли.
2. Избыток андрогенов пренатального периода. Наиболее важной проблемой, вызванной накоплением предшедственников стероидных гормонов, является сбрасывание 17-гидроксипрогестерона в путь биосинтеза андрогенов, что приводит к высокому уровню андростендиона, который превращается вне надпочечников в тестостерон. Эта проблема возникает у пораженных плодов к 8-10 нед гестации и приводит к аномальному развитию половых органов у девочек (см. рис. 1, 2).
Наружные половые органы у мальчиков и девочек в норме приобретают половую принадлежность в течение внутриутробного периода. Девочки с этим нарушением, в/утробно подвергающиеся воздействию высоких уровней андрогенов надпочечникового происхождения, имеют маскулинизированные наружные половые органы (см. рис. 1, 2), что проявляется увеличением клитора, частичным/полным сращением половых губ. Влагалище обычно имеет общее отверстие с уретрой (урогенитальный синус). Клитор может быть увеличен настолько, что напоминает половой член; поскольку отверстие уретры располагается ниже этого органа, некоторые девочки, страдающие данным нарушением, могут ошибочно считаться мальчиками с гипоспадией и крипторхизмом.
Выраженность вирилизации обычно сильнее у девочек с сольтеряющей формой дефицита 21-гидроксилазы (см. табл. 2). Внутренние половые органы нормальные, т.к. у девочек, страдающих этим нарушением, имеются нормальные яичники, а не яички, и поэтому не секретируется антимюллеров гормон.
Пренатальное воздействие высоких уровней андрогенов на ГМ может влиять на последующий половой диморфизм поведения у девочек с этим нарушением. Девочки могут демонстрировать агрессивную модель поведения, интересоваться игрушками для мальчиков, напр. машинками и грузовиками, и часто мало заинтересованы в игре с куклами. Женщины могут быть мало заинтересованы в роли матери. Высока частота гомосексуальности у девушек, страдающих этим нарушением. Тем не менее большинство из них действуют гетеросексуально и не имеют сомнений в половой принадлежности/дисфории. Женщины с этим нарушением обычно не присваивают себе мужскую роль, за исключением некоторых из них с самой тяжелой степенью вирилизации.
Младенцы мужского пола выглядят нормальными при рождении. Таким образом, диагностика у мальчиков м.б. не проведена до тех пор, пока не появятся признаки недостаточности надпочечников. В связи с тем, что состояние пациентов с этим нарушением может быстро ухудшиться, младенцы мужского пола умирают чаще, чем младенцы женского пола. По этой причине во всех 50 американских штатах и во многих др. странах проводится неонатальный скрининг на наличие этого состояния.
3. Избыток андрогенов постнатального периода. У детей обоего пола, получающих неадекватное/не получающих лечение, развиваются дополнительные признаки избытка андрогенов после рождения. Диагностика у мальчиков с простой вирильной формой дефицита 21-гидроксилазы обычно отсрочена, т.к. они выглядят нормальными, у них редко развивается недостаточность надпочечников.
Признаки избытка андрогенов включают быстрый соматический рост, ускоренное созревание скелета. Т.о., пациенты с этим нарушением имеют высокий рост в детстве, однако преждевременное закрытие эпифизов вызывает относительно раннюю остановку роста, и во взрослом возрасте они низкорослы (см. рис. 1). Развитие мускулатуры м.б. избыточным. Может появиться оволосение лобковой области, подмышечных впадин, могут формироваться акне, низкий голос. Половой член, мошонка, простата у мальчиков с этим нарушением могут увеличиваться; однако яички имеют допубертатный размер, поэтому могут казаться маленькими по сравнению с увеличенным половым членом. Иногда происходит гиперплазия эктопических кл. коры надпочечников в яичках пациентов, подобно аналогичным в надпочечниках, из которых образуются опухоли яичек из эктопической ткани надпочечника. У девочек с этим нарушением клитор в дальнейшем может увеличиваться (см. рис. 1).
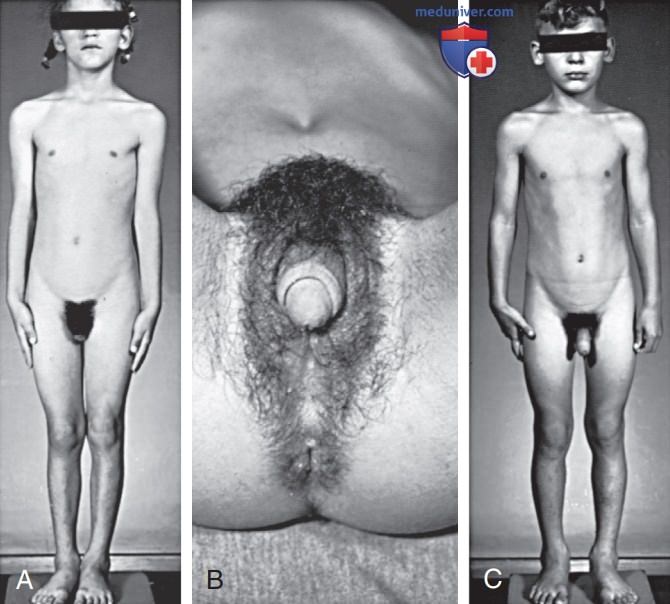
Рисунок 1. А — 6-летняя девочка с вирилизирующей врожденной гиперплазией надпочечников. Рост соответствовал возрасту 8,5 лет, костный возраст составлял 13 лет; В — обратите внимание на увеличение клитора и сращение половых губ; С — родители не считали, что у ее 5-летнего брата имеются нарушения. Рост соответствовал возрасту 8 лет, костный возраст составлял 12,5 лет
Несмотря на то, что внутренние половые органы являются женскими, развития груди, менархе может не произойти, если избыточная продукция андрогенов не будет подавлена адекватным лечением.
При неклассической форме дефицита 21-гидроксилазы возникают сходные, но менее выраженные признаки избытка андрогенов (см. табл. 2). При этой мягкой форме отмечаются нормальные уровни кортизола, альдостерона, половые органы у девочек с этим нарушением после рождения нормальные. У мальчиков и девочек может наблюдаться преждевременное половое созревание, раннее развитие оволосения лобковой области, подмышечных впадин. В более позднем периоде м.б. гирсутизм, акне, нарушения менструального цикла, бесплодие, но большинство девочек и мальчиков имеют полностью асимптоматическое течение.
4. Дисфункция мозгового вещества надпочечников. Развитие мозгового в-ва надпочечников требует воздействия экстремально высоких уровней кортизола, что происходит в надпочечниках в норме. У пациентов с классической врожденной гиперплазией надпочечников функция мозгового в-ва надпочечников м.б. аномальной, о чем свидетельствуют ослабленные адреналиновые реакции, гипогликемия, ЧСС при ФН. Способность к физическим упражнениям не нарушена, но клиническое значение этих данных неясно. Дисфункция мозгового в-ва надпочечников может усугублять действие дефицита кортизола на ССС у пациентов, получающих неадекватное/не получающих лечение.
д) Лабораторные признаки. См. табл. 1.
Пациенты с сольтеряющей формой заболевания имеют характерные лабораторные данные, ассоциированные с дефицитом кортизола и альдостерона, включая гипонатриемию, гиперкалиемию, метаболический ацидоз, гипогликемию, но развитие этих отклонений после рождения может занять >10-14 дней. Уровни 17-гидроксипрогесте-рона в крови повышены. Однако уровни этого гормона высоки в течение первых 2-3 дней после рождения даже у младенцев, не страдающих этим нарушением, особенно если они больны/недоношены. По окончании периода младенчества, как только установится циркадианный ритм кортизола, уровни 17-гидроксипрогестерона изменяются в том же циркадианном ритме (самые высокие утром, самые низкие ночью). У пациентов с сольтеряющей формой заболевания уровни кортизола в крови низкие.
У пациентов с простой вирильной формой заболевания они обычно нормальные, но непропорционально низкие относительно уровней АКТГ и 17-гидроксипрогестерона. Кроме 17-гидроксипрогестерона, у девочек, страдающих этим нарушением, также повышены уровни андростендиона и тестостерона; у мальчиков уровень тестостерона не повышен, т.к. у здоровых младенцев мужского пола имеются высокие уровни тестостерона, сравнимые с таковыми у детей более старшего возраста. Уровни 17-кетостероидов и прегнантриола в моче повышены, но это исследование редко применяется в клинической практике, т.к. проще получить образцы крови, чем собрать суточную порцию мочи.
Уровни АКТГ повышены, но по сравнению с уровнями 17-гидроксипрогестерона не имеют диагностического значения. Уровни ренина в плазме крови повышены, сывороточного альдостерона — непропорционально низки относительно уровня ренина. Однако у здоровых детей в первые несколько недель жизни отмечается высокий уровень ренина.
Наиболее точно диагноз дефицита 21-гидроксилазы можно установить путем измерения уровня 17-гидроксипрогестерона до и спустя 30 или 60 мин после в/в болюсного введения 0,125-0,25 мг козинтррпина (АКТГ 1-24). Существуют номограммы, которые позволяют легко отличать здоровых лиц, пациентов с неклассическим и классическим дефицитом 21-гидроксилазы. У гетерозиготных носителей этого АуР-заболевания уровни АКТГ-стимулированного 17-гидроксипрогестерона более высоки, чем у лиц без генетических нарушений, но существует значительное совпадение между субъектами из этих двух категорий. У младенцев с явными электролитными нарушениями/нестабильностью кровообращения проведение этого исследования м.б. невозможным/для этого потребуется отложить лечение, пока в случайном образце крови уровни предшественников не будут достаточно повышены для проведения диагностики.
В клинических условиях доступно проведение генотипирования, что может помочь подтвердить диагноз, но это дорого, может занять несколько недель. Поскольку конверсии генов, которые формируют большинство мутантных аллелей, могут передавать более одной мутации, должно быть также проведено генотипирование по крайней мере одного родителя, чтобы определить, какие мутации находятся на каждом аллеле.
е) Дифференциальная диагностика (ДД). Нарушения полового развития обсуждаются более подробно в отдельной статье на сайте - просим Вас пользоваться формой поиска по сайту выше. Начальным этапом оценки младенца с половыми органами промежуточного типа является тщательное физикальное обследование для описания анатомии половых органов, нахождения наружного отверстия мочеиспускательного канала, пальпации мошонки/половых губ и паховой области на предмет наличия яичек (пальпируемые половые железы почти всегда указывают на наличие ткани яичек и, следовательно, на то, что младенец имеет мужской генотип), поиска любых др. анатомических аномалий. УЗИ может помочь увидеть наличие/отсутствие мочеиспускательного канала, определить расположение половых желез.
Быстрое кариотипирование (например, флуоресцентная гибридизация интерфазных ядер X- и У-хромосом in situ) может помочь быстро определить генетический пол младенца.
Все эти данные, вероятно, будут доступны до получения результатов гормонального исследования и в совокупности позволят команде клиницистов консультировать родителей относительно генетического пола ребенка, анатомии внутренних репродуктивных органов. Введение контрастной среды в урогенитальный синус девочки с вирилизацией позволяет увидеть влагалище и матку, поэтому большинство хирургов используют эти данные, чтобы составить план хирургического ведения.
ж) Пренатальная диагностика. Проведение пренатальной диагностики дефицита 21-гидроксилазы возможно на поздних сроках I триместра методом анализа ДНК, полученной при биопсии ворсин хориона/во время II триместра при помощи амниоцентеза. Обычно это делается потому, что у родителей уже есть ребенок, страдающий этим нарушением. Чаще всего проводят анализ гена CYP21 на наличие наиболее часто возникающих мутаций; более редкие мутации можно определить при помощи ДНК-секвенирования.
з) Неонатальный скрининг. В связи с тем, что у мальчиков дефицит 21-гидроксилазы часто не диагностируется до появления признаков выраженной недостаточности надпочечников, во всех американских штатах и во многих др. странах проводятся программы неонатального скрининга. В соответствии с этими программами проводится анализ уровня 17-гидроксипрогестерона в сухой капле крови, полученной при заборе крови из пятки с последующей абсорбцией на фильтровальном бумажном тест-бланке; эти же тест-бланки параллельно проверяются на наличие других врожденных заболеваний: гипотиреоза, фенилкетонурии. В отношении детей с потенциально возможным нарушением обычно быстро проводят дополнительное исследование (определение электролитов, повторное определение 17-гидроксипрогестерона) в возрасте ~2 нед.
У младенцев с сольтеряющей формой заболевания к этому возрасту часто имеются электролитные нарушения, но они обычно не являются тяжелобольными. Т.о., программы скрининга во многих случаях эффективны для предупреждения адреналового криза у мальчиков, страдающих этим нарушением. Неклассическая форма заболевания достоверно не выявляется при неонатальном скрининге, но это имеет небольшое клиническое значение, поскольку при данном типе дефицита 21-гидроксилазы недостаточность надпочечников не возникает.
Основная трудность современных программ неонатального скрининга заключается в очень высокой частоте ложноположительных результатов, т.к. для достоверного выявления всех младенцев с данным нарушением в качестве минимального диагностически значимого результата устанавливается крайне низкий уровень 17-гидроксипрогестерона (т.е. тест имеет низкую положительную прогностическую ценность — 1%). У недоношенных детей дело обстоит еще хуже. Положительная прогностическая ценность может быть увеличена, если минимальный диагностически значимый результат будет основан на гестационном возрасте, а также с помощью более специфических методов скрининга 2-го уровня (жидкостная хроматография с последующей тандемной масс-спектрометрией).
и) Лечение:
1. Заместительная терапия глюкокортикостероидами. ГКС в больших дозах, чем при др. формах недостаточности надпочечников, — ЛП выбора для лечения дефицита кортизола, подавления избыточной продукции андрогенов корой надпочечников, что минимизирует проблемы с чрезмерным ростом, созреванием скелета, вирилизацией — 15-20 мг/м2/сут гидрокортизона ежедневно внутрь Q8H. У младенцев с данным нарушением обычно требуется применение дозировок, находящихся в верхней части этого диапазона (в РФ: 10-15 мг/м2/сут, а на первом году жизни до 20 мг/м2/сут)*.
P.S. * Федеральные КР (протоколы) по ведению детей с эндокринными заболеваниями.Под ред. И.И. Дедова, В.А. Петерковой. М.: Практика, 2014.
Применение двойных/тройных доз показано во время стрессовых периодов, например инфекций/операций. Лечение ГКС должно быть продлено на неопределенный срок у всех пациентов с классическим дефицитом 21-гидроксилазы, может не требоваться пациентам с неклассической формой заболевания при отсутствии признаков избытка андрогенов. Лечение должно быть индивидуальным. Желательно поддерживать линейный рост в пределах процентильных линий; переход к более высоким процентилям роста может указывать на недостаточное лечение, тогда как снижение процентилей роста часто указывает на передозировку ГКС. Передозировку также можно предположить в случае чрезмерного набора веса. Необходимо наблюдать за половым развитием, проводя периодические осмотры; созревание скелета оценивается при помощи серийных рентгеновских снимков кисти и запястья для оценки костного возраста.
Измерение уровня гормонов, особенно 17-гидроксипрогестерона, андростендиона, необходимо проводить ранним утром до приема утренних ЛП/в определенное время, привязанное к приему лекарств. Желательные уровни 17-гидроксипрогестерона находятся в диапазоне высоконормальных значений/в несколько раз выше нормы; низконормальные уровни обычно достигаются только при передозировке ГКС. Проведены небольшие клинические исследования ЛП с альтернативными формами доставки, включая таблетки гидрокортизона с замедленным высвобождением и использование п/к-устройства для непрерывной инфузии инсулина (инсулиновой помпы) для доставки гидрокортизона по схеме, более близкой к нормальному суточному ритму секреции кортизола. На данный момент эти подходы еще не внедрены в клиническую практику.
Менархе наступает в соответствующем возрасте у большинства девочек, у которых удалось достичь хорошей компенсации; у девочек с субоптимальной компенсацией его наступление м.б отсрочено. Диагностика у детей с простой вирильной формой заболевания м.б. не проведена до 3-5-летнего возраста, когда степень зрелости скелета может опережать календарный возраст на >5 лет. У некоторых детей, особенно если костный возраст соответствует >12 годам, может возникнуть спонтанное центральное (т.е. гонадотропин-зависимое) половое созревание при проведении лечения, т.к. терапия гидрокортизоном подавляет продукцию андрогенов надпочечниками, стимулируя высвобождение гонадотропинов гипофизом, если гипоталамус достиг соответствующей степени зрелости. Лечение этой формы сочетанного истинного преждевременного полового созревания можно проводить аналогом гонадотропин-рилизинг-гормона, таким как лейпролид.
У мальчиков с дефицитом 21-гидроксилазы, которым проводилась неадекватная терапия ГКС, могут развиться опухоли яичка из эктопической ткани надпочечников, которые обычно регрессируют при увеличении доз стероидов. МРТ, УЗИ, цветное допплеровское картирование яичек помогают определить характер и степень заболевания. Сообщалось о щадящей операции на яичках при опухолях, не реагирующих на терапию ГКС.
2. Заместительная терапия минералокортикоидами. Пациентам с сольтеряющей формой заболевания (т.е. дефицитом альдостерона) требуется заместительная терапия флудрокортизоном. Младенцы могут иметь очень высокие потребности в минералокортикоидах в 1-й мес жизни (0,1-0,3 мг/сут Q12H, иногда до 0,4 мг/сут). Часто в дополнение к минералокортикоиду требуется восполняющее введение натрия (натрия хлорида, 8 ммоль/кг). Поддерживающая доза флудрокортизона для более старших младенцев и детей — 0,05-0,1 мг/сут. У некоторых пациентов ремиссию простой вирильной формы заболевания легче поддерживать низкими дозами флудрокортизона в сочетании с гидрокортизоном, даже если уровни альдостерона у этих пациентов нормальные без заместительной терапии минералокортикоидами.
Терапия оценивается путем мониторинга жизненно важных показателей; тахикардия, гипертензия — признаки передозировки минералокортикоидов. В раннем младенческом возрасте при подборе терапии требуется проводить частые измерения уровня электролитов сыворотки крови. Определение активности ренина плазмы крови — эффективный способ оценки адекватности терапии; она должна поддерживаться в пределах/вблизи нормального диапазона, но не подавляться.
Были предложены дополнительные подходы по улучшению исхода, однако пока они не вошли в стандарты оказания МП: применение антиандрогенов (флутамид для блокирования действия избыточных уровней андрогенов) и/или ингибитора ароматазы (анастрозол, который блокирует превращение андрогенов в эстрогены, тормозя созревание скелета — процесс, чувствительный к эстрогенам, как у мальчиков, так и у девочек). Ингибиторы ароматазы в целом не должны применяться у девушек в период полового созревания в связи с замедлением естественного процесса полового созревания, чрезмерным воздействием гонадотропинов на яичники. Для улучшения роста во взрослом возрасте было предложено применение гормона роста с агонистами люлиберина, замедляющими процесс созревания скелета, или без них.
3. Хирургическое лечение половых органов промежуточного типа. Девочкам с выраженной вирилизацией операция проводится в возрасте между 2 и 6 мес жизни. При наличии выраженной клиторомегалии проводят уменьшение клитора с частичным удалением тела органа, с сохранением нервно-сосудистого пучка; однако умеренная клиторомегалия может стать менее заметной даже без хирургического вмешательства по мере роста пациентки. Во время операции на клиторе обычно проводится пластика влагалища и коррекция урогенитального синуса; в подростковом возрасте часто необходима ревизия.
С родителями девочек с этим нарушением необходимо подробно обсудить риски и пользу хирургического лечения. За функциональными исходами у пациенток, перенесших современные хирургические процедуры, проводится долгосрочное наблюдение в течение ограниченного срока. Судя по всему, частота и выраженность сексуальной дисфункции возрастает у девушек с наиболее значительными степенями генитальной вирилизации и с наиболее выраженной степенью ферментативных нарушений (пренатальное воздействие андрогенов), вызванных мутациями у каждой пациентки (см. табл. 2). Выбор пола младенцам с нарушениями половой дифференцировки (включая врожденную гиперплазию надпочечников) основан на ожидаемом половом функционировании, фертильности во взрослом возрасте с ранней хирургической коррекцией наружных половых органов в соответствии с избранным полом.
Расстройство гендерной идентичности при этом заболевании встречается нечасто; оно возникает в основном у девочек с сольтеряющей формой гиперплазии надпочечников, наиболее выраженной степенью вирилизации.
Противники генитальной хирургии при других расстройствах половой дифференцировки из непрофессиональных сообществ и медицинских кругов высказывают озабоченность тем, что такое вмешательство игнорирует необъективность ролевой гендерной предрасположенности, определенной пренатально воздействием андрогенов, и исключает возможность принятия пациентом какого-либо решения относительно предпочитаемой им самим гендерной идентичности и того, какую хирургическую коррекцию гениталий следует проводить. Они настаивают на том, что лечение должно быть направлено в первую очередь на просвещение пациента, семьи и др. людей о патологическом состоянии, на его лечение и на то, как жить с состоянием интерсексуальности.
Они предлагают отложить операцию до тех пор, пока пациент не решит, какую коррекцию следует провести. Не все немедицинские сообщества поддерживают идею отсроченного хирургического лечения, многие согласны с необходимостью проведения операции в младенческом возрасте. Генотипические (XX) девочки с выраженной вирилизацией, которые росли как мальчики, в целом хорошо себя ощущают в роли мужчин во взрослом возрасте.
У девушек-подростков и взрослых женщин с плохо контролируемым дефицитом 21-гидроксилазы (гирсутизм, ожирение, аменорея) альтернативой стандартной лекарственной заместительной гормональной терапии может служить двусторонняя лапароскопическая адреналэктомия (с заместительной гормональной терапией), однако в связи с удалением надпочечников пациенты, лечение которых проводится таким способом, имеют наиболее высокий риск возникновения острой недостаточности надпочечников в случае прерывания лечения. Более того, у них могут проявляться признаки повышения уровня АКТГ (патологическая пигментация).
4. Лечение в пренатальном периоде. Помимо генетического консультирования, основная цель пренатальной диагностики — облегчение пренатального лечения девочек с гиперплазией надпочечников. Беременным из группы риска рождения ребенка с этим нарушением можно назначать дексаметазон — стероид, легко проникающий через плаценту, — ежедневно 20 мкг/кг веса матери до беременности Q8-12H для подавления секреции стероидов надпочечниками плода, включая секрецию адрогенов. Если терапия начата к 6 нед гестации, то она поможет снизить степень вирилизации наружных половых органов у девочек с этим нарушением. Затем для определения пола, генотипа плода проводится биопсия ворсин хориона; терапию продолжают только в том случае, если плод оказывается девочкой, у которой имеется данное нарушение.
Анализ ДНК клеток плода, отделенных от материнской плазмы, для определения пола и анализ гена CYP21 могут позволить на ранних этапах выявить наличие данного нарушения у плода женского пола. Лечение следует рассматривать только у страдающих данным нарушением плодов женского пола. Дети под влиянием этой терапии имеют немного меньший вес при рождении. Предполагалось наличие влияния на личность и мышление, например появление повышенной застенчивости, однако четкой связи не наблюдалось. Имеющейся на данный момент информации недостаточно для того, чтобы определить, допустимо ли наличие долгосрочных рисков, особенно у мальчиков и девочек, не страдающих данным нарушением, которые не получают прямой пользы от лечения. Побочные эффекты пренатального лечения со стороны матери включали отеки, чрезмерный набор веса, гипертензию, нарушение толерантности к глюкозе, кушингоидный тип лица, выраженные стрии.
В совместном заключении профессиональных сообществ рекомендовано проводить пренатальное лечение только в рамках протоколов исследования, но в некоторых регионах оно предлагается как вариант лечения вне условий клинических исследований акушерами, ведущими беременности высокой степени риска.