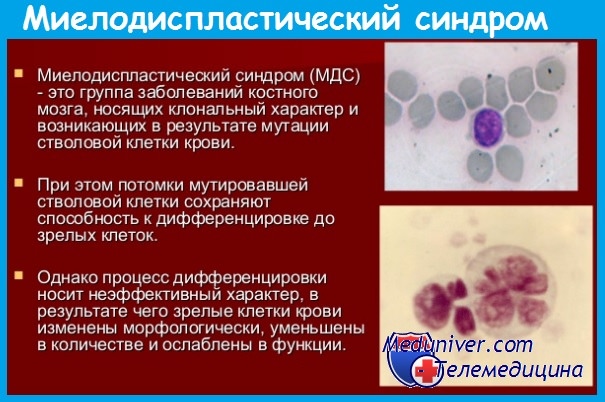
Клональность миелодиспластических синдромов (МДС) - клональная теория
При миелодиспластических синдромах, как и при лейкозах, клоновый характер кроветворения подтверждается обнаружением во всех клетках крови одного типа изофермента глюкозо-6-фосфатдегидрогеназы (Г-6-ФД) у женщин, гетерозиготных по этому энзиму; в то время как гемопоэтические элементы у них содержат только один тип Г-6Ф-Д, в остальных клетках, в частности в фибробластах, определяются оба типа энзима.
Клоновый характер кроветворения подтверждается и цитогенетическими исследованиями, в том числе методом FISH (флюоресцентной гибридизацией in situ), позволяющим обнаруживать хромосомные аберрации в клетках как в метафазе, так и в интерфазе, методом RFLP (оценкой случайной ингибиции одной из Х-хромосом), а также исследованием онкогенов, включая их точечные мутации и реаранжировки (перестройки и утраты).
Имеются доказательства того, что первично при миелодиспластических синдромах (МДС) поражается стволовая кроветворная клетка. Это положение подтверждается выявлением изофермента А Г-6ФД у женщин, гетерозиготных по этому энзиму, с диагнозом «рефрактерная анемия с кольцевыми сидеробластами», не только в клетках миелоидного, но и лимфоидного ростков кроветворения.
Другим доказательством служит обнаружение одинаковых хромосомных аберраций в клетках миелоидной линии, В-лимфоцитах и стволовых клетках. Например, делеция 5q (5q-) была выявлена методом FISH не только в клетках миелоидной направленности дифференцировки, но и в лимфоидных клетках с про-В-иммунофенотипом (CD34+, CD19+), а также в стволовых клетках (CD34+, CD38).
Молекулярно-биологические методы исследования указывают на наличие мутации гена NRAS в гранулоцитах, моноцитах, Т- и В-лимфоцитах, а также генов, расположенных на хромосоме X лимфоцитов и кодирующих гипоксантинфосфорибозилтрансферазу (HPRT), а также фосфоглицераткиназу (PGK). Исследования полиморфизма гена, расположенного на хромосоме X, кодирующего рецептор андрогена у человека (HUMARA), также подтвердили вовлечение в клоновый процесс лимфоцитов.
Кроме того, о поражении стволовой клетки свидетельствует возможность трансформации заболевания не только в острый миелоидный, но и, крайне редко, — в острый лимфобластный лейкоз, преимущественно В-клеточной направленности дифференцировки. Описано несколько случаев ОЛЛ с характерными иммунологическими (TdT+, CD19+, CD10+) и цитогенетическими маркерами — t(4;11) или Ph'+(p190bcr-abl).
Кроме того, J. F. Leseve и соавт. описали два случая рефрактерной анемии с избытком бластов без трансформации в острый лейкоз с наличием хромосомы Ph' и белка p190bcr-abl. В обзоре, опубликованном Т. J. Hamblin, указывается на 14 наблюдений билинейного и 7 — бифенотипического острого лейкоза, развившихся из миелодиспластического синдрома.
Однако существуют данные, указывающие на возможность первичного поражения при миелодиспластических синдромах (МДС) не стволовой клетки, а раннего миелоидного предшественника. Это предположение подтверждают исследования клеток крови и костного мозга больных миелодиспластическими синдромами (МДС), имеющих хромосомные аберрации, выявленные методами FISH и ПЦР (полимеразная цепная реакция). Например, по данным некоторых авторов, трисомия хромосомы 8 (+8) и моносомия хромосомы 7 (-7) были выявлены в клетках миелоидного ростка кроветворения, но отсутствовали в лимфоцитах.
Высказывается мнение, что описанные нарушения кариотипа, возможно, появляются на более позднем этапе болезни в результате дополнительных мутаций в миелоидном субклоне. Тем не менее современные методы исследования позволяют более убедительно говорить о происхождения патологического клона из стволовой клетки, что постулируется классификацией ВОЗ в отношении всех вариантов миелодиспластических синдромов (МДС).