Мутация гена CSF1R (синоним FMS) выявляется в 12—20 % случаев миелодиспластических синдромов (МДС), особенно при вариантах с увеличенным числом бластных клеток. Этот ген в норме кодирует рецептор макрофагального колониестимулирующиего фактора. Мутантный ген часто связан с неблагоприятным прогнозом как в отношении трансформации миелодиспластических синдромов (МДС) в ОМЛ, так и низкой выживаемости больных.
Мутация гена FLT3 (представленная внутренней тандемной дупликацией) диагностируется приблизительно в 10 % случаев миелодиспластических синдромов (МДС) и ОМЛ с трехростковой дисплазией. Этот ген кодирует III класс рецептора тирозинкиназы, участвующий в пролиферации и дифференцировке стволовых клеток. Мутация гена FLТЗ ассоциирована с трансформацией миелодиспластических синдромов (МДС) в ОМЛ и низкой продолжительностью жизни больных.
Гиперэкспрессия гена С-KIT, ответственного за синтез рецептора стволово-клеточного фактора, особенно характерна для вариантов миелодиспластических синдромов (МДС) с увеличенным числом бластных клеток в костном мозге.
Гиперэкспрессия гена MPL, кодирующего рецептор тромбопоэтина, встречается преимущественно у больных миелодиспластическими синдромами (МДС) с увеличенным числом бластных клеток и ассоциирована с неблагоприятным прогнозом, включая высокую вероятность трансформации миелодиспластических синдромов (МДС) в ОМЛ.
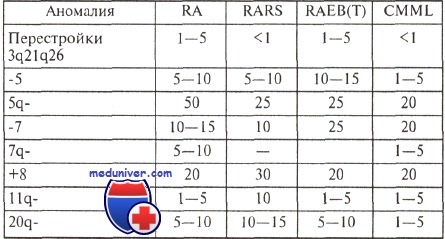
Частота (в процентах) характерных аномалий кариотипа при различных миелодиспластических синдромах
Точечная мутация гена GCSFR, вызывающая дефект рецептора G-CSF (гранулоцитарный колоние-стимулирующий фактор), является возможным объяснением развития миелодиспластических синдромов (МДС) и ОМЛ у больных с тяжелой врожденной нейтропенией (severe congenital neutropenia). Однако для возникновения миелодиспластических синдромов (МДС) или ОМЛ одной мутации гена GCSFR недостаточно. Развитие этих заболеваний ассоциировано с моносомией хромосомы 7 и мутацией гена RAS.
Мутации генов семейства RAS (наиболее часто гена NRAS) встречаются, по данным разных исследований, в 10—40 % случаев миелодиспластических синдромов (МДС), чаще при нарушениях миеломоноцитарной дифференцировки, например при хроническом миеломоноцитарном лейкозе. Мутация и гиперэкспрессия гена NRAS, участвующего в трансдукции сигнала с поверхности клетки, ассоциированы с повышенным риском развития ОМЛ и, возможно, являются одним из первых этапов лейкемической трансформации миелодиспластических синдромов.
Мутация гена опухолевой супрессии ТР53 (синоним Р53), кодирующего белок Р53, отмечается в 5— 25 % всех случаев миелодиспластических синдромов, особенно часто при вторичном характере МДС. Ген ТР53 в норме обеспечивает контроль целостности генома, и в случае повреждения ДНК либо происходит «арест» клеточного цикла в фазах G1 и G2, ли о активация апоптоза клеток. Существуют данные об увеличении частоты мутации гена ТР53 при «продвинутых» ФАБ-вариантах МДС — РАИБ и РАИБ-Т, а также при трансформации миелодиспластических синдромов (МДС) в ОМЛ.
Ген AML1 (синоним RUNX1) участвует в регуляции дифференцировки клеток миелоидного ростка кроветворения. Его мутация относится к одной из наиболее часто встречающихся мутаций при ОМЛ. При миелодиспластических синдромах мутация гена AML1 обнаруживается редко — в 5 % случаев.
Исследование Н. Harada и соавт. указывает на сопоставимую с данными предыдущей работы частоту мутации AML1 в случайной выборке больных МДС — 2,7 % и значительно более высокую — 46 % среди больных, подвергшихся воздействию малых доз облучения (выживших после атомной бомбардировки Хиросимы), а также — у 38 % больных вторичными миелодиспластическими синдромами и ОМЛ, ранее получавших терапию алкилирующими препаратами с локальным лучевым лечением и без такового лечения.
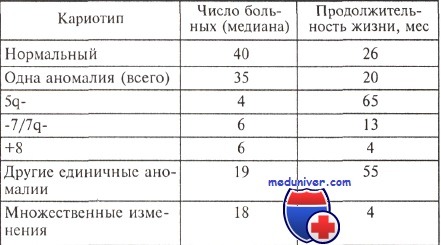
Продолжительность жизни больных миелодиспластическим синдромом с различными изменениями кариотипа
В патогенезе миелодиспластических синдромов задействовано множество других генов: реаранжировка гена MLL выявлена у 47 % больных, повышенная экспрессия гена WT1 — у 44 %, гена HFE — y 50 % [440], а также гена DLK1. Выявлены отсутствие или выраженное уменьшение экспрессии гена DCC.
При миелодиспластических синдромах (МДС), помимо мутаций генов, обнаружено гиперметилирование ДНК — присоединение метальной группы к цитозину. В настоящее время выявлено гиперметилирование четырех генов, регулирующих клеточный цикл: Р15INK4b, Р16INK4a, Р14ARF и гена ретинобластомы.
При миелодиспластических синдромах (МДС) наиболее изученным является ген р15INK4b. Его гиперметилирование отмечается у 26—63 % больных, причем не только в бластных клетках, но и в гранулоцитах крови. Гиперметилирование этого гена чаще определяется при вариантах МДС с увеличенным числом бластных клеток костного мозга (48 % больных), чем при вариантах с их нормальным числом (30 % больных).
Определенное внимание в патогенезе миелодиспластических синдромов уделяется повышенной активности теломеразы — рибонуклеиновому ферменту, содержащему фрагмент РНК, комплементарный ДНК теломеры (последовательности нуклеотидов, завершающих конец хромосом, которые стабилизируют их и предотвращают от разрушения). Повышенная активность теломеразы наряду с укорочением теломер может быть ранним маркером генетической нестабильности, приводящей в дальнейшем к возникновению хромосомных аберраций.
Точечная мутация митохондриальной ДНК и/или РНК может быть ранним этапом в патогенезе миелодиспластических синдромов. Мутации митохондриальной ДНК были выявлены в 40-60% случаев миелодиспластических синдромов. При рефрактерной анемии с кольцевыми сидеробластами выявлена мутация гена COII (митохондриальной ДНК) и генов 4MTIS1 и 4MTTD (митохондриальной тРНК).