Гипопроконвертинемия — геморрагический синдром, возникающий по поводу недостатка синтеза Ф. VII, характеризующийся в клиническом отношении тяжелыми провоцированными геморрагиями. Существуют врожденная форма и приобретенная форма.
Обособленная с нозологической точки зрения в рамках группы геморрагических диатезов с удлиненным TQ, врожденная форма считалась весьма частым заболеванием; в дальнейшем было установлено, что под названием гипопроконвертинемии существовало в дейтсвительности два порока, которые разделились на: недостаток Ф. VII и недостаток Ф. X.
После переоценки случаев в свете вышеуказанного, констатировалось, что это заболевание возникает намного реже, причем частота его равняется 0,1/100 000. Среди животных это заболевание встречается у собак породы Бигль ("Beagle").
Первый случай описал и изучил Alexander в 1951 г., откуда данному заболеванию было присвоено имя «парагемофилия Александера»; до 1979 г. в литературе опубликовано 85 случаев.
Чистая приобретенная форма до сих пор не упоминалась в литературе по специальности. Сообщается лишь о случаях приобретенного недостатка Ф. VII в ассоциации с недостатком Ф. II, IX и X, в обстоятельствах упомянутых при недостатке Ф. II (авитаминоз К) и недостатков Ф. II, V, IX, X при заболеваниях печеночной паренхимы.
Патофизиология гипопроконвертинемии
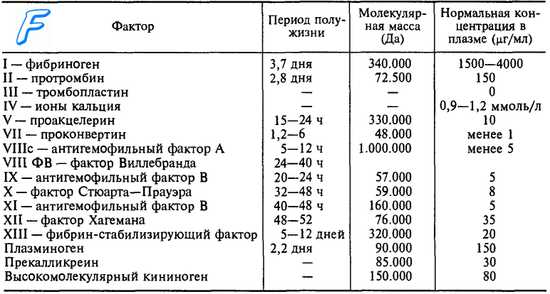
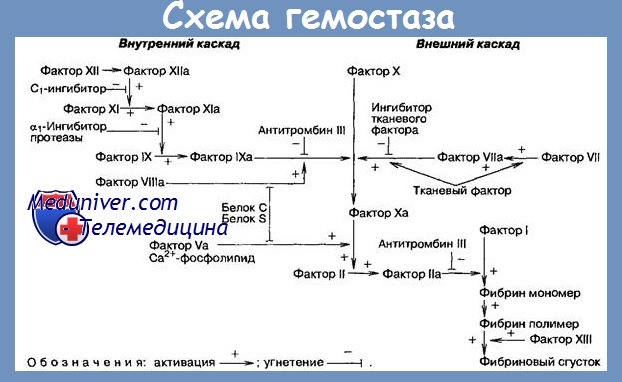
Основное расстройство гипопроконвертинемии — недостаток синтеза Ф. VII, который у нормального лица выполняется гепатоцитами и регулируется парой генов, находящихся на соматических хромозомах. У больных, ингибиция этих генов имеет своим результатом недостаточный синтез Ф. VII, ниже необходимого уровня для гемостаза (25%), что порождает геморрагический диатез.
Генетическая передача автосомально-рецессивного характера и ее фенотипические выражения подобны тем, которые существуют при недостатке Ф. V. Этот недостаток обычно семейный; он затрагивает в одинаковой мере и мальчиков и девочек.
Гомозиготы, благодаря затронутости обоих генов (Ф. VII = 0—5%) представляют клинические признаки болезни; гетерозиготы, благодаря затронутости только одного гена (Ф. VII = 30—50%) являются здоровыми носителями порока и выявляются лишь по случаю лабораторного исследования.
Приобретенная форма, в сочетании с недостатками других плазматических факторов коагуляции, встречается в этиопатогенетических обстоятельствах, подобных тем, которые мы наблюдаем при недостатке Ф. П.