Данная группа включает состояния с анатомическими и/или функциональными признаками поражения базальных структур, отвечающих за контроль движений. Изменения могут ограничиваться экстрапирамидной системой или сочетаться с более диффузным поражением или нарушением функций. Аномалии движений являются основным проявлением. Наиболее частым нарушением движений у детей являются тики, но только в некоторых случаях они представляют тяжелые и стойкие патологические изменения. Дистония является наиболее типичным нарушением движений, требующим обследования и лечения (Fernandez-Alvarez и Aicardi, 2001). Акинетико-ригидный синдром редко встречается у детей.
Состояния, при которых отсутствуют определенные патологические признаки поражения базальных ганглиев, но отмечаются аномалии движений (то есть тики, синдром Туретта, тремор, зеркальные движения) кратко описаны в конце данного раздела.
Хорея представлена тремя основными заболеваниями. Острые хореи, в частности, хорея Сиденгама, рассмотрены в отдельных статьях на сайте. В этой сттатье представлены хорея Гентингтона, хорео-акантоз и доброкачественная наследственная хорея. Хорея также может быть проявлением нескольких заболеваний, некоторые из которых поддаются лечению.
а) Хорея Гентингтона. Хорея Гентингтона является генетическим заболеванием, наследуемым по доминантному типу и характеризующимся экстрапирамидными проявлениями, которые могут быть достаточно вариабельными и включают несколько типов аномальных движений, среди которых хорея является классической формой заболевания у взрослых, а гипокинетическо-ригидный синдром — наиболее типичной формой в детском возрасте; также характерна прогрессирующая деменция.
Заболевание связано с распространением повторов тринуклеотида ЦАГ внутри кодирующего региона гена, картированного на дистальном конце 4р хромосомы. Продукт гена (гантингтин) является новым белком с неизвестной функцией. В контрольной группе выявляется менее 30 повторов; среди пациентов с хореей — 36-121 триплетов (в среднем 44) (Kremer et al., 1994). В соответствии с генетическим механизмом отмечается антиципация (прогрессирующее более раннее проявление симптомов в ряду поколений) и связь наследования с полом. Ювенильные формы, в частности ригидная форма, наследуются от отца в 70-90% случаев (Но et al., 2001). Длина повторов тринуклеотида приблизительно в трети случаев существенно увеличивается при наследовании. Отмечается положительная корреляция раннего начала и большей тяжести заболевания. Распространение происходит внутри полиглутаминового пути и приводит к образованию внутриклеточных включений, и в некоторой степени приводит к усилению токсического действия.
Хорея Гентингтона редко встречается в детском возрасте, несмотря на то, что среди населения в целом составляет 1 на 24000, а распространенность — 7,61 на 100000. Частота встречаемости гетерозигот может составлять 1 на 5000. Тем не менее, случаи начала заболевания до 20-летнего возраста составляют только лишь 5-10% от общего числа пациентов. Вероятно, внешние факторы оказывают некоторое влияние на возраст начала и выраженность заболевания (van Dellen и Hannan, 2004).
С точки зрения патологии отмечается выраженная атрофия стриатума, в особенности хвостатого ядра, приводящая к исчезновению нормального выступа головки хвостатого ядра в передние рога желудочков. Также отмечается атрофия лобной коры. Выявляется истощение нейронов, затрагивающее преимущественно мелкие нейроны, являющиеся ГАМК-эргическими, но могут содержать и субстанцию Р и энкефалины. Крупные нейроны, содержащие преимущественно соматостатин или нейропептид Y, поражаются только в тяжелых случаях. Содержание липофусцина в стриатуме повышено, а само вещество уплотнено, в некоторых случаях отмечается дегенерация митохондрий. Снижение количества клеток связано со снижением содержания ГАМК и синтеза ацетилхолина, обнаруживаемого на основании низкого уровня ацетилхолин-трансферазы. Концентрация соматостатина и пептида Y также снижена. Концентрация дофамина внутри стриатума не отличается от нормы, а введение леводопы приводит к усилению хореиформных движений.
В редких случаях заболевание встречается у детей, когда у родителей еще не развились клинические проявления. Из 46 зарегистрированных случаев начала заболевания в детстве средний возраст появления симптомов составил 3-9 лет (Osborne et al., 1982). Наиболее частым проявлением у детей является ригидность, но несмотря на это расстройство интеллекта может предшествовать появлению экстрапирамидных симптомов. По результатам нейропсихологического исследования у взрослых с доклинической стадией заболевания выявляется раннее поражение функций лобной доли. По данным одного из исследований, ригидный ювенильный вариант болезни Гентингтона отмечался у 26 из 46 пациентов, хореиформные движения — у 9 пациентов, а сочетание хореи и ригидности — у 3 пациентов (в обзоре Osborne). Хореиформные движения сходны с проявлениями хореи Сиденгама, тем не менее, они могут затрагивать в большей степени туловище и проксимальную часть конечностей. Дизартрия может быть ранним и преобладающим симптомом и проявлением поражения мозжечка.
Тем не менее, для детского возраста гораздо более типичен паркинсонизм с ригидностью, чем хореиформные движения. Часто заметна двигательная апраксия. Припадки отмечаются у 50% больных детей (Osborne et al., 1982), также описана миоклоническая форма заболевания (Gambardella et al., 2001). На ЭЭГ могут выявляться ритмичные затылочные медленные волны в сочетании со спайками (Ullrich et al., 2004). Биохимические маркеры заболевания отсутствуют, таким образом, диагностика зависит от клинических проявлений и семейного анамнеза.
При визуализации выявляется расширение желудочков с атрофией головки хвостатого ядра. При ригидных формах может отмечается усиление сигнала от стриатума. Изменения МРТ могут быть поздним проявлением, а локализация снижения метаболизма, выявляемая при позитронно-эмиссионной томографии с помощью деоксиглюкозы, является более чувствительным методом обследования (Berent et al., 1988). Выявление распространенности повторов тринуклеотида ЦАГ подтверждает диагноз. Тем не менее, недавно у взрослых были зарегистрированы редкие случаи болезни, подобной болезни Гентингтона (Stevanin et al., 2003). В некоторых случаях избыток тринуклеотида ЦАГ не отмечается (Vuillaume et al., 2000); в других случаях заболевание связано с распространением мутации ЦТГ/ЦАГ (Margolis et al., 2004).
Дифференциальная диагностика включает редкие случаи младенческого паркинсонизма, лекарственную и спонтанную дистонию и аномальные движения, болезнь Халлерфордена-Шпатца, болезнь Вильсона, дистонический липидоз, синдром Леша-Найхана, хорею Сиденгама, стриато-таламическую дегенерацию (Gieron et al., 1995), доброкачественную наследственную хорею и хореоакантоз в молодом или даже подростковом возрасте. Последнее заболевание проявляется как хореиформными движениями, так и периферической нейропатией с амиотрофией нижних конечностей и когнитивными и поведенческими нарушениями. Акантоциты выявляются в крови. В некоторых случаях наследственная хорея без деменции может отмечаться у пациентов с распространением повторов тринуклеотида ЦАГ, что предполагает широкий диапазон фенотипической вариабельности болезни Гентингтона (Britton et al., 1995). Отмечается прогрессирующая атрофия головки хвостатого ядра и аномальный сигнал от лентикулярного ядра. При патологоанатомическом исследовании выявляется атрофия таламуса и стриатума.
Возможна доклиническая и даже пренатальная ДНК диагностика болезни Гентингтона. Данное обстоятельство может быть источником этических проблем, но позволяет родителям принять осознанное решение.
Эффективного лечения в настоящее время не существует. Галоперидол уменьшает аномальные движения. Некоторое положительное действие могут оказывать диазепам, тетрабеназин, натрия вальпроат и карбамазепин. Исследуются новые терапевтические методики, включая перенос генов и новые препараты (Hannan 2004; Melone et al., 2005).
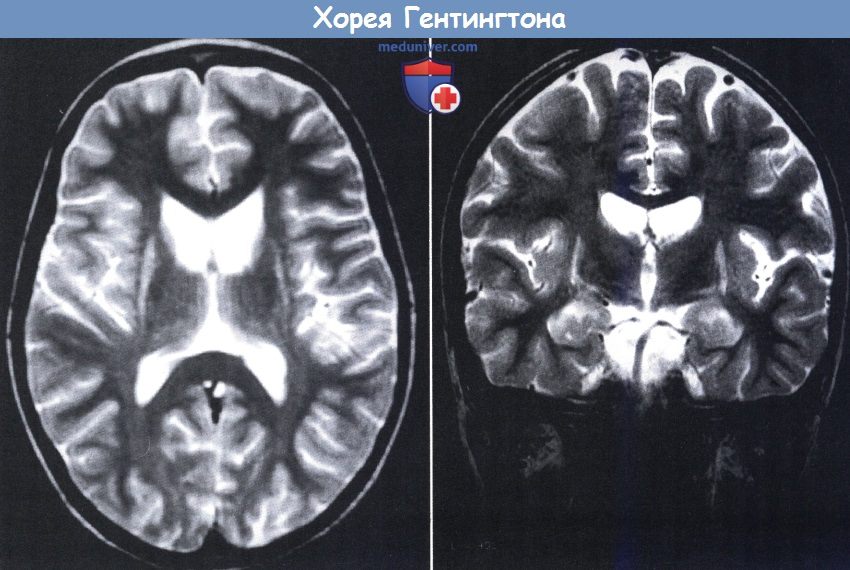
Хорея Гентингтона. четырехлетняя девочка с припадками, миоклонией и деградацией развития.
В аксиальной (слева) и фронтальной (справа) проекциях на МРТ видна атрофия хвостатого ядра с результирующим увеличением передних рогов и усилением сигнала от внешней скорлупы.
б) Доброкачественные наследственные хореи. Несколько лет известны очень редкие доброкачественные формы хронической хореи без когнитивной деградации. Заболевания гетерогенны по клиническим и генетическим изменениям (Kleiner- Fisman et al., 2003), таким образом, существование доброкачественной хореи спорно. Отмечаются изолированные хореические движения без когнитивных или неврологических нарушений. Зарегистрировано небольшое число семей с соответствующей клинической картиной изолированной непрогрессирующей хореи, начинающейся в детском возрасте с нормальными когнитивными функциями (Breedveld et al., 2002а); обнаружены мутации гена TITF-1 на 14 хромосоме (Breedveld et al., 2002b). Мутации были выявлены только в 3 из 10 обследованных семей, что свидетельствует о генетической гетерогенности заболевания. Случаи, не связанные с 14 хромосомой, характеризуются более вариабельным фенотипом с дистонией и/или миоклонусом, тремором или нейросенсорной тугоухостью (Wheeler et al., 1993), заболевание может прогрессировать.
Данный вариант течения может представлять собой несколько отдельных заболеваний. В некоторых случаях отмечается тенденция к уменьшению выраженности хореических движений с возрастом, то есть у больных родителей симптомы могут иметь неявный характер. В одной семье зарегистрировано состояние, описанное как «обратная хорея», при котором в первую очередь и в большей степени поражены нижние конечности (Fisher et al., 1979). В некоторых случаях явной доброкачественной хореи отмечается увеличение количества повторов тринуклеотида ЦАГ, сходное с признаками хореи Гентингтона, тем не менее, чаще всего отмечается относительно небольшое число повторов (36-39) (Britton et al., 1995). Такие изменения могут оставаться бессимптомными, но при этом передаваться следующему поколению в более тяжелой форме, поэтому оправдана молекулярная диагностика. Диагноз основан на семейном анамнезе и отсутствии когнитивных нарушений. В целом, лечение не требуется, отмечается тенденция к уменьшению выраженности заболевания с течением времени.
в) Хорея-акантоцитоз (нейроакантоцитоз). Нейроакантоцитоз — генетическое заболевание, относящееся наряду с синдромом МакЛеода, болезнью Гентингтона 2, ассоциированной с пантотенат-киназой нейродегенерацией (PKAN) и абеталипопротеинемией к группе редких заболеваний, характеризующихся наличием в крови шпоровидных эритроцитов (Rampoldi et al., 2002). С неврологической точки зрения часто отмечается поражение базальных ганглиев и периферических нервов в сочетании с дискинезиями, ухудшением когнитивных функций, психиатрическими нарушениями и прогрессирующим течением (Danek et al., 2005). Абеталипопротеинемия относится к группе спиноцеребеллярных атаксий, a PKAN — к дистониям.
Нейроакантоцитоз является редким рецессивным нейродегенеративным заболеванием, поражающим преимущественно взрослых, тем не менее, отмечены случаи болезни у подростков и детей. Заболевание является результатом мутации гена VPS13A, функция которого неизвестна. Основным клиническим проявлением являются нарушения движений, представленные хореей и дистонией; особенно выражено поражение лица и области щек, часто в виде тиков. Часто встречаются дизартрия и дисфагия, когнитивные нарушения и психиатрические проявления, утрата сухожильных рефлексов и другие проявления сенсомоторной нейропатии. Аномалии движений глаз включают частые вертикальные подергивания и медленные гипометрические саккады, в особенности при взоре вверх (Gradstein et al., 2005). Заболевание начинается незаметно, и первые нехарактерные проявления могут предшествовать основным симптомам и развиваться за 2-20 лет до формирования полной клинической картины (Lossos et al., 2005).
Диагноз может быть подтвержден исследованием ДНК и при обнаружении в крови белка хореина (Dobson-Stone et al., 2004). Зарегистрированы также случаи доминантного наследования (Walker et al., 2002).
Неврологический фенотип синдрома МакЛеода сходен с хорея-акантоцитозом, но наследование сцеплено с Х-хромосомой (Danek et al., 2001). Лицевые тики и дизартрия встречаются реже, а кардиомиопатия — чаще, чем при рецессивных формах. Характерным является отсутствие Кх-антигена и низкий уровень антигена Kell в крови. Диагноз подтверждается при выявлении мутации гена YX на Х-хромосоме. В редких случаях возможно появление симптомов у женщин с пораженным геном. Проводится только симптоматическое лечение.