Первичная DYT1 дистония в большинстве случаев вызвана мутациями гена DYT1 на хромосоме 9q34. Тем не менее, данные мутации, включающие 80% случаев заболевания среди евреев-ашкенази (Bressman, 2004), гораздо реже обнаруживаются среди лиц нееврейской национальности. Из 150 пациентов европейского происхождения, страдающих идиопатической дистонией, у 22 выявлялась данная мутация, а у 15-19 отмечался типичный фенотип с началом в виде поражения конечностей, в то время как из 128 пациентов без мутации гена DYT1 у 46 отмечалась сегментарная, а у 59 очаговая дистония, у 23 — генерализованная форма, имитирующая фенотип DYT1 (Valente et al., 1998). Мутация гена DYT1 представлена в большинстве случаев парной делецией трех оснований ЦАГ и обнаруживается как у евреев ашкенази, так и у лиц нееврейской национальности, но в первой группе встречается значительно чаще. Продуктом гена является белок, называемый торсин А1, роль которого до конца не изучена.
Генетические предпосылки торсионной дистонии достаточно хорошо изучены: заболевание наследуется доминантным путем, характеризуется сниженной пенетрантностью (20-40%), среди носителей гена встречается множество неполных форм. Тем не менее, часто выявляются формы заболевания, не сцепленные с 9ql4, большая часть таких случаев имеет спорадический характер. Нередко встречаются атипичные и неполные формы. В данном случае установленный риск для родственников первой линии составляет 21%, у 75% носителей мутантного гена симптомы появляются до 30 лет (Fletcher et al., 1990). При наличии мутации гена DYT1 возможна пренатальная диагностика, но в связи с относительно низкой пенетрантностью и вариабельной выраженностью симптомов предсказать фенотип невозможно.
Идиопатическая торсионная дистония не сопровождается патологоанатомическими изменениями. Функциональное поражение базальных ганглиев можно предположить на основании аномалии распределения и метаболизма нейротрансмиттеров, включающей снижение уровня норадреналина и дофамина, а также пептидэргического нейротрансмиттера соматостатина.
Клинические проявления варьируют. Заболевание обычно начинается после пяти лет, но возможно и более ранее начало, включая редкие случаи появления симптомов на первом году жизни (Mostofsky et al., 1996). Примерно в половине случаев симптомы появляются до 15 лет. Среди пациентов препубертатного возраста в 85% случаев генетически подтвержденного заболевания первый симптом затрагивал нижние конечности (Bressman, 2004); конечности поражались у 95% пациентов, в то время как туловище и шея только у 25-38% пациентов. Тем не менее, процент пациентов нееврейского происхождения с поражением нижних конечностей был примерно одинаковым по результатам некоторых исследований (Angelini et al., 1988).
Дистония изначально имеет очаговый или сегментарный характер. Типичная ранняя картина заболевания включает сгибание стопы. В большинстве случаев нарушение походки имеет нетипичный и интермиттирующий характер, что приводит к ошибочной диагностике истерии. В начале заболевания дистония часто зависит от выполняемого действия. Некоторые дети могут нормально выполнять нетипичные движения (например, ходить задом наперед, в то время как контрактура становится явной при движении вперед). Эмоциональное напряжение и стресс приводят к усилению дистонии. В 70% случаев заболевания, начавшегося до 11 лет, отмечается генерализация. В итоге пораженная конечность или конечности принимают более или менее постоянное аномальное положение с наслаивающимися изменениями, которые также поражают оставшиеся части тела. Часто встречается лордоз, распространено латеральное изгибание туловища с торсией вдоль вертикальной оси.
В большинстве случаев лицо не поражено, а речь и глотание нарушены только у небольшого количества пациентов. В редких случаях выявляется висцеральная дистония с затруднением дыхания. Несмотря на то, что моторные нарушения могут препятствовать любым видам произвольных движений и даже угрожать жизни, интеллект всегда сохранен.
Дистония с поздним началом развивается у подростков и молодых людей. В таких случаях отмечается тенденция к локализованной дистонии или ограниченному распространению заболевания. Среди пациентов старше 15 лет обычно поражаются шея (пароксизмальная кривошея) и верхние конечности. Зарегистрирована дистония, ограничивающаяся одной мышечной группой (Stojanovic et al, 1995).
Писчий спазм является нетипичным начальным проявлением, но другие дистонии при целенаправленных действиях (например, при игре на музыкальном инструменте) могут быть первым проявлением (Stojanovic et al, 1995). Формы с поздним началом обычно протекают более легко и склонны к сегментарным проявлениям.
Отмечается тенденция к стабилизации течения через несколько лет. Окончательный исход при детских формах достаточно плохой, так как половина пациентов прикована к постели или инвалидному креслу к 35 годам, в то время как при позднем начале заболевания только в редких случаях невозможно самостоятельное передвижение. Течение заболевания обычно, но не всегда, прогрессирующее, также отмечаются некоторые изменения интенсивности, особенно при позднем начале. Неожиданное обострение симптомов, известное как дистонический статус, встречается редко, но угрожает жизни и требует экстренного лечения (Manji et al., 1998; Nardocci et al., 2005).
Атипичные варианты классической торсионной дистонии редко встречаются среди евреев ашкенази, но часто выявляются в других этнических группах. Атипичные формы включают краниоцервикальное поражение, подобные миоклонусу движения, формы с первичным поражением осевой мускулатуры, а не конечностей, а также легкие и очаговые формы, выявляемые после определения взаимосвязи с типичными формами. Зарегистрировано пять случаев, когда начало заболевания имело явную связь с травмой или введением галоперидола, и в одном случае основным проявлением являлись бульбарные знаки (Edwards et al., 2003). Тремор может сочетаться с идиопатической дистонией. Спазматическая дисфония, вызванная дистонией приводящих мышц голосовых связок, встречается редко. В некоторых тяжелых случаях развиваются длительные и интенсивные спазмы, которые могут угрожать жизни.
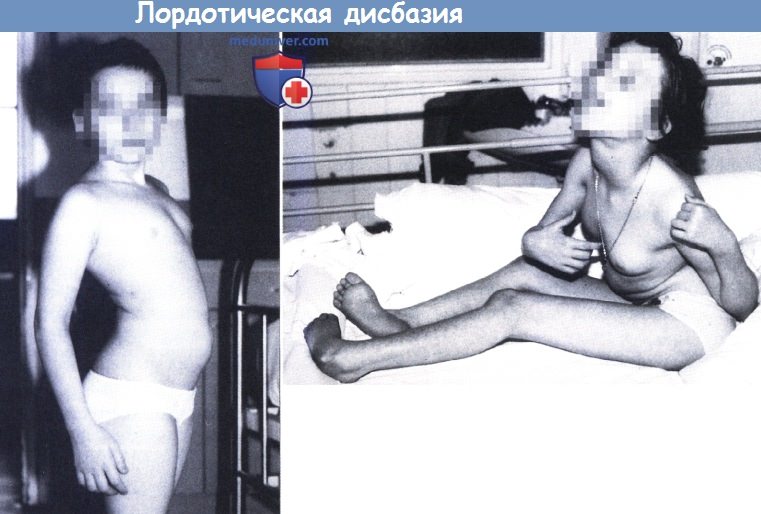
Лордотическая дисбазия. Начальные проявления у семилетней девочки.
Отмечается заметный лордоз (слева). Тяжелая форма заболевания у восьмилетней девочки.
Отмечается искривление стопы, деформация позвоночника, дистоническое положение правой руки и ретроколлис.
У данной пациентки также выявлено заметное поражение мышц гортани и глотки и непрерывное выталкивание языка (справа).
а) Диагностика первичной торсионной дистонии. Важной целью диагностических исследований является исключение вторичных дистоний, особенно поддающихся лечению. МР-визуализация необходима для исключения органических заболеваний, которые могут имитировать первичную дистонию. Функциональная визуализация для идентификации дофаминовых рецепторов представляет в большей степени патофизиологический интерес. То же касается и других лабораторных исследований, результат которых в случае DYT1 всегда отрицательный.
Дифференциальная диагностика включает истерию, которая часто ошибочно диагностируется в связи с атипичными и вариабельными проявлениями при нормальных результатах визуализации и лабораторных исследований. Наиболее важными для дифференциальной диагностики в связи с возможностью лечения являются болезнь Вильсона и допа-зависимая дистония, также следует думать о дискинетическом церебральном параличе и других частично излечимых заболеваниях, включая дефицит креатинина или редкое заболевание базальных ганглиев, чувствительное к биотину (Ozand et al., 1998). Исследование уровня леводопы должно проводиться во всех случаях, когда отсутствуют признаки других заболеваний.
Медикаментозная дистония, которая может развиться при приеме антипсихотических или противосудорожных препаратов, может сочетаться с другими дискинетическими проявлениями. Отсроченная дистония у детей с пренатальными или приобретенными повреждениями базальных ганглиев может возникать до 5-10-летнего возраста (Saint-Hilaire et al., 1991). Наличие аномалий при визуализации является диагностическим критерием. Атипичные позы, напоминающие дистонию, могут встречаться при грыжах пищеводного отверстия диафрагмы (синдром Сандифера). Данное состояние важно иметь в виду, так как возможно хирургическое лечение рефлюксной болезни.
б) Другие наследственные первичные дистонии. Наиболее важной формой является DYT5, известная как допа-зависимая дистония, которая изначально может проявляться изолированным дистоническим фенотипом. Другие формы изолированной дистонии встречаются редко и чаще всего регистрируются в особых этнических группах или только в небольшом количестве семей. Они включают миоклоническую дистонию и редкие заболевания, среди которых в настоящее время выделено 13 различных генетических форм (de Carvalho Aguiare и Ozelius, 2002). Большая часть других форм зарегистрирована в небольшом количестве семей или в специфических этнических группах (Fernandez-Alvarez и Aicardi, 2001; Bressman, 2004; Fasano et al., 2005) или в связи со специфическим хромосомным локусом.
DYT2 тип описан среди цыган (Khan et al., 2003); ген DYT4 обнаружен в сочетании с «шепчущей дистонией» (de Carvalho Aguiare и Ozelius, 2002); ген DYT6 сцеплен с 8-й хромосомой (Almasy et al., 1997); ген DYT7 обнаружен в небольшом количестве семей в Германии и сочетается с очаговой дистонией с началом во взрослом возрасте (Leube et al., 1997); ген DYT13, расположенный на 1р36 хромосоме может сочетаться с мио-клоническими проявлениями (Bentivoglio et al., 2004). Клинические различия, выявляемые в некоторых этнических группах (например, среди шведов и франкоканадцев), предполагают гетерогенность заболевания. Сцепленные с Х-хромосомой формы обычно проявляются дистонией-паркинсонизомом (Kupke et al., 1990), а измененный ген картирован на Хц13-хромосоме.
в) Транзиторная идиопатическая дистония у младенцев. Транзиторные дистонические симптомы, не связанные с какими-либо аномалиями развития или неврологическими отклонениями, возникают у некоторых детей в возрасте старше пяти лет (Willemse et al, 1986). Транзиторная идиопатическая дистония младенцев проявляется в возрасте от пяти месяцев до одного года и не имеет явных причин. Заболевание проявляется аномальным положением тела, обычно в области одной или обеих рук и характеризуется приведением или пронацией со сгибанием запястья. В редких случаях дистония может затрагивать туловище или нижние конечности в определенных положениях и утихает в период сна. Дистонические положения исчезают, когда ребенок совершает целенаправленные движения пораженной конечностью.
Описанные аномалии обычно исчезают к концу первого года (Fernandez-Alvarez и Aicardi, 2001). Данное доброкачественное состояние обычно не имеет генетических предпосылок, несмотря на ряд зарегистрированных семейных случаев. Зарегистрированы случаи пароксизмальной дистонии у младенцев в виде кратких эпизодов опистотонуса и дистонии верхних конечностей с благоприятным исходом (Angelini et al., 1988).