Ассоциированная с пантотенат киназой-2 дегенерация (PKAN; болезнь Галлервордена-Шпатца)
Несмотря на то, что данное заболевание может быть причиной нескольких типов нарушений движений, оно описано в данном разделе в связи с тем, что дистония является преобладающим симптомом.
Заболевание характеризуется следующими патологоанатомическими изменениями:
1) наличием ржаво-коричневого железно-кальциевого пигмента в паллидуме и черной субстанции,
2) симметричным разрушением паллидума и решетчатой части черной субстанции,
3) наличием сфероидов в базальных ганглиях и (в меньшей степени) в коре (Dooling et al, 1974).
Данный патологический синдром неспецифичен, и некоторые исследователи относят его к группе заболеваний, называемых нейродегенерациями с накоплением железа в головном мозге; данная группа заболеваний также включает нейроаксональную дистрофию и ацерулоплазминемию и может отмечаться при некоторых определенных энцефалопатиях (Aicardi и Castelein, 1979; Hartig et al., 2006). С другой стороны, PKAN является прогрессирующим дегенеративным рецессивно наследуемым заболеванием с упомянутыми выше патологическими изменениями. Несмотря на то, что заболевание часто рассматривается как форма младенческой нейроаксональной дистрофии (болезнь Зайтельбергера), недавно была доказана его генетическая обособленность (Hortnagel et al., 2004).
Мутантный ген PKAN2 находится на хромосоме 20р12.3.
Ассоциированная с пантотенат киназой нейродегенерация (PKAN).
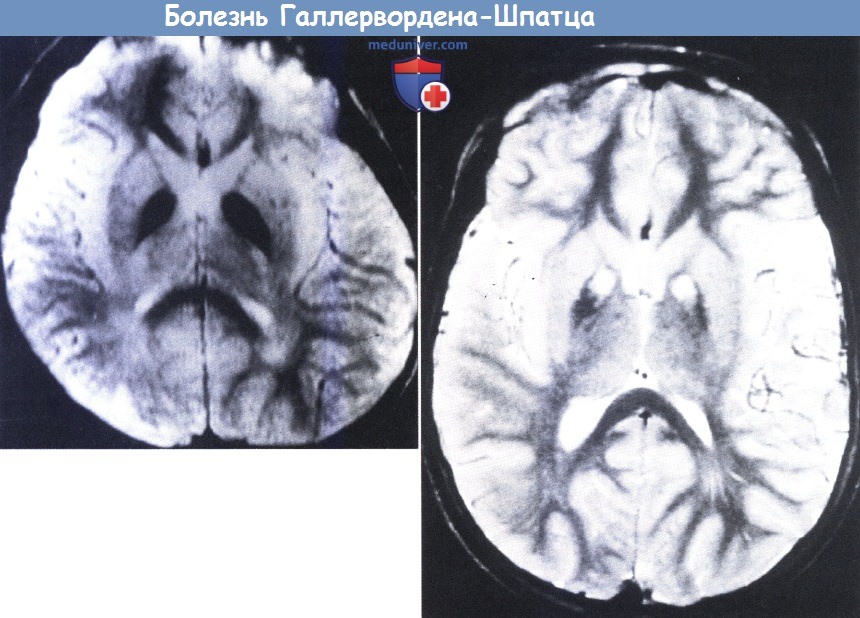
На МРТ (слева) видно заметное снижение сигнала от паллидума, вероятно связанное с содержанием железа.
На МРТ другого пациента (справа) виден центр темного паллидума, симметричные участки высокого сигнала в Т2-режиме, так называемый признак «глаз тигра».
Данные участки соответствуют некрозу и могут отсутствовать в начале заболевания, что продемонстрировано на рисунке слева.
Рано начинающаяся форма заболевания манифестирует в младенчестве в виде медленного развития и часто изначально кажется статичной. Выявляются двусторонние пирамидные знаки, что вместе с медленным течением часто вызывает подозрение на наличие непрогрессирующего заболевания. Фаза регресса наступает в возрасте 5-10 лет и проявляется исчезновением двигательных и речевых навыков, преобладающей дистонией в области рта и нижней челюсти (Savoiardo et al., 1993) и поведенческими нарушениями. Значимость когнитивной деградации варьирует. Часто вместе с поведенческими нарушениями отмечается хореоатетоз с последующей интеллектуальной деградацией.
Среди симптомов ювенильной формы преобладает прогрессирующая дистония, приводящая к формированию фиксированных дистонических поз. Заболевание начинается в возрасте 6-28 лет. Дистонические движения отмечаются в области лица и рта. У трети пациентов формируется стагнация или регресс когнитивного развития. Уже на ранних стадиях приблизительно у половины пациентов выявляется ретинопатия, также может отмечаться акантоцитоз. На МРТ выявляются характерные изменения в виде низкого сигнала в Т2-режиме с участками высокого сигнала, вероятно, соответствующими некротическим тканям, так называемые повреждения по типу «глаза тигра» (Angelini et al., 1992; 0stergaard et al., 1995).
Тем не менее, данные изменения могут иметь только транзиторный характер или не выявляются в некоторых случаях, когда отмечается только снижение плотности. Гипобеталипопротеинемия может отмечаться в сочетании с акантоцитозом и пигментным ретинитом, данное сочетание зарегистрировано при отдельном синдроме (гипобеталипопротеинемия, акантоцитоз, пигментный ретинит или синдром HARP). Данные проявления могут отмечаться только при особой форме заболевания (Higgins et al., 1992). Приблизительно у трети пациентов формируются поведенческие и интеллектуальные нарушения, а течение заболевания прогрессирующее. Описаны формы заболевания, сопровождающиеся атрофией зрительного нерва (Casteels et al., 1994). Недавно стало уделяться особое внимание значимой вариабельности фенотипа и генотипа (Pellecchia et al., 2005), заболевание является генетически гетерогенным.
Отмечены случаи без мутации гена PKAN2, они могут отличаться от классических форм отсутствием ретинопатии и наличием атрофии зрительного нерва, изменения при визуализации не отличаются (Hartig et al., 2006). Диагностика основана на клинических проявлениях и изменениях на МРТ, диагноз подтверждается генетическими исследованиями. Исход заболевания неблагоприятный, у пациентов быстро развивается тяжелая инвалидность. В некоторых случаях эффективна стимуляции паллидума (Castelnau et al., 2005). В настоящее время лечение отсутствует, за исключением таких проявлений как дистония и ригидность.