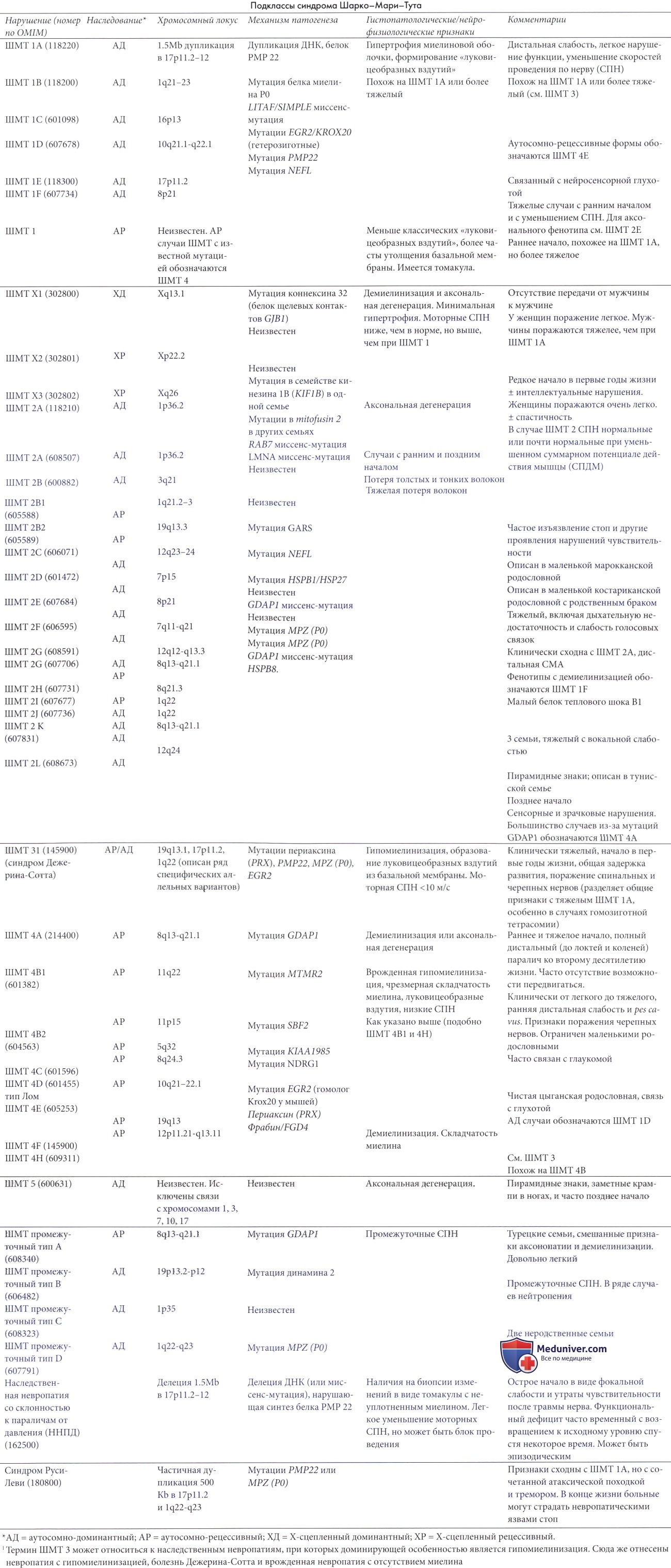
Синдром Шарко-Мари-Тута 3 (ШМТ 3, НСМН 3 типа, болезнь Дежерина-Сотта)
Термины ШМТ 3, НСМН III типа и болезнь Дежерина-Сотта относятся к гетерогенной группе наследственных или спорадических невропатий (Ouvrier et al., 1987, 1999) с вариантами возраста начала и тяжести. Возраст начала значительно отличался у двух пациентов, исходно описанных Дежерином и Сотта.
Отсутствует единое мнение относительно границ или даже существования этой группы, особенно для врожденных типов, но по нашему мнению, она остается полезным клиническим понятием. Была выявлена значительная генетическая гетерогенность. Сообщалось о мутациях следующих видов: мутации и делеции, часто сочетанные, генов MPZ, РМР22, периаксина, и EGR2, гомозиготность по генотипу ШМТ 1А с или без дупликации и гомозиготность по синдрому ШМТ 2, вызванному мутацией миелина Р0| (Sghirlanzoni et al., 1992; Tyson et al., 1997).
С патоморфологической точки зрения изменения нерва напоминают таковые при типе 1, но они более интенсивны. Происходит выраженное уменьшение миелинизированных волокон, особенно толстых. Луковицеобразные вздутия хорошо сформированы, но часто состоят из пластинок удвоенных базальных мембран, и соотношение диаметра аксона с общим диаметром волокна выше нормального, что указывает на гипомиелинизацию (Ouvrier et al., 1987). В некоторых случаях имеется массивная интерстициальная коллагеновая гипертрофия.
а) Классический фенотип. При наиболее типичной форме (Ouvrier et al., 1987) начало раннее, с замедлением развития моторики и гипотонией, обычно проявляющимися в течение первого года жизни. Способность ходить отсрочена у 30-50% пациентов. Слабость более глубокая и диффузная, чем в случаях 1 типа и часто охватывает проксимальные мышцы. Последовательно появляется атаксия, и слабость может быть асимметричной. Экзофтальм с утолщением и выворотом краев — весьма частый симптом, в особенности при мутациях РМР22 (Ouvrier et al., 1987).
В некоторых случаях наблюдаются зрачковые нарушения и даже синдром Аргайлла Робертсона. У пожилых пациентов сухожильные рефлексы отсутствуют, и гипертрофия нервов является частой.
Течение заболевания тяжелее, чем при ШМТ 1 и ШМТ 2, но в небольшой степени, хотя некоторые пациенты неспособны к самостоятельной ходьбе к пубертатному периоду.
Скорости проведения по нерву значительно уменьшаются до уровня ниже 10 м/с или даже не могут быть измерены из-за отсутствия вызванной стимуляцией реакции мышцы. У родителей нет никаких клинических симптомов или уменьшения СПН, что таким образом исключает возможность невропатии 1 типа, но сообщается, что иногда ЭМГ демонстрирует признаки легкого неврогенного поражения мышц.
б) Врожденные демиелинизирующие невропатии. Врожденные демиелинизирующие невропатии обычно рассматривают как форму ШМТ 3, хотя раннее начало встречается и в некоторых случаях ШМТ 1 (Harati и Butler, 1985). Они также составляют гетерогенную группу, по поводу нозологии которой единое мнение отсутствует.
Guzzetta et al. (1982) различали две группы врожденных случаев. Тяжелая форма может напомнить острую болезнь Верднига-Гоффманна с врожденной гипотонией и параличом, атрофией и нарушением дыхания, которое может стать летальным через несколько месяцев или лет (Goebel et al., 1976) усугубляясь нарушениями глотания.
Повышенное содержание белка в ЦСЖ является постоянным, и скорости проведения по нерву не поддаются измерению или чрезвычайно низки. Существуют менее тяжелые случаи, которые позволяют жить на протяжении многих лет, но с тяжелой двигательной недостаточностью. В ходе таких случаев может наблюдаться некоторая сенсорная недостаточность.
Патоморфологическая картина в большинстве случаев представлена полным отсутствием миелина (безмиелиновая форма или невропатия с отсутствием миелина) (Ouvrier et al., 1999) с формированием луковицеобразных вздутий, содержащих, главным образом, слои шванновских клеток с малым количеством или отсутствием миелина. Молекулярно-биологическая основа случаев с отсутствием миелина определена недостаточно.
Патоморфологические данные интерпретировались как подтверждение того, что процесс является скорее недостаточностью депонирования миелина, чем демиелинизацией-ремиелинизацией. В некоторых случаях наряду с демиелинизацией присуствует и пролиферация микрофиламентов в шванновских клетках (Ulrich et al., 1981). В других был описан нестабильный миелин с образованием складок миелиновых пластинок (Peudenier et al., 1993). Сообщалось о случаях с врожденным артрогрипозом (Balestrini et al., 1991; Boylan et al., 1992).
Johnson et al. (1989) описали центральную демиелинизацию в сочетании с демиелинизирующей полиневропатией. Похожие случаи были вызваны мутацией SOX10 в сочетании с болезнью Гиршпрунга (Inoue et al., 2002) и мутациями GJA12, ответственными за аутосомно-рецессивную форму болезни типа Пели-цеуса-Мерцбахера (Uhlenberg et al, 2004).
Gharndi et al. (1997) и Levy et al. (1997) описали два необычных случая восстановления после врожденной демиелинизирующей невропатии.