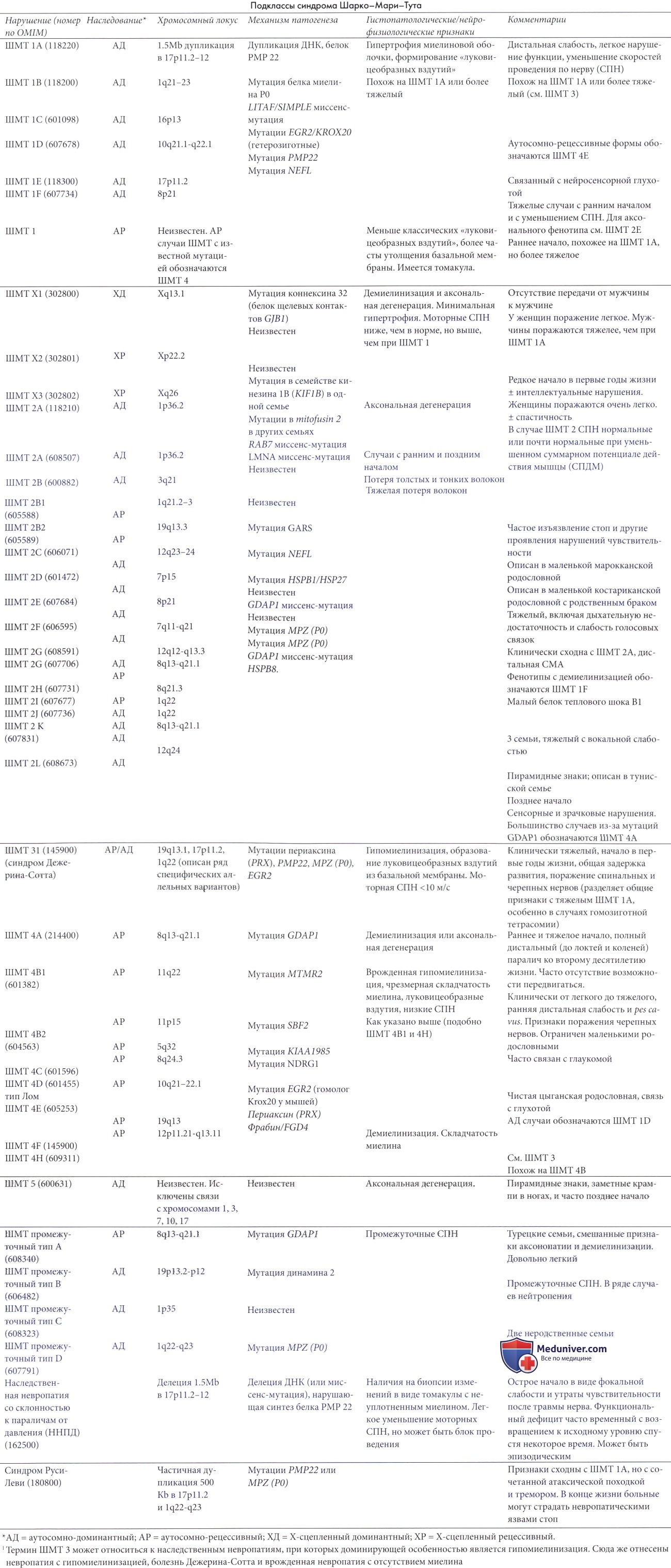
Синдром Шарко-Мари-Тута (ШМТ) 2 также вызывает клиническую картину, согласующуюся с описанием болезни Шарко-Мари-Тута. Ее частота у детей традиционно намного ниже, чем в случае болезни Шарко-Мари-Тута (ШМТ) 1 (Ouvrier 1992; Ouvrier et al., 1999; Ouvrier и Wilmhurst 2003). Высокая распространенность болезни среди шведских детей может быть следствием этнических факторов; однако, частота бессимптомной или очень легкой формы у маленьких детей осложняет диагностику и отчасти объясняет противоречивые данные.
а) Патоморфология. Патоморфология характеризуется наличием признаков аксональной дегенерации с вторичным поражением миелиновой оболочки или шваннновских клеток. Уменьшается количество миелинизированных волокон, особенно большого диаметра, могут наблюдаться кластеры регенерации. Луковицеобразные вздутия не являются частыми. Участки нервного волокна между перехватами Ранвье укорочены и имеют неравномерную длину при препарировании иглой. Острые аксональные поражения редки, и патоморфологические аномалии могут быть незаметными.
б) Клиническая картина. Клинические проявления подобны таковым при болезни Шарко-Мари-Тута (ШМТ) 1, но часто имеют более позднее начало, в течение второго или третьего десятилетия, и более медленное течение. Атрофия задней большеберцовой и икроножной мышц обычно столь же заметна, как и в переднелатеральном компартменте. Отсутствие рефлексов и деформация стопы не столь часты, как при типе 1, но могут встречаться язвы на ноге, позволяя выделить определенные подтипы, такие как ШМТ 2В, трудно отличимый от наследственной сенсорной и вегеативной невропатии (НСВН) типа 1 (Elliott et al., 1997).
Сенсорную симптоматику часто трудно продемонстрировать. В данном случае отсутствует гипертрофия нерва, и увеличение белка в ЦСЖ встречается редко. Тремор верхних конечностей нетипичен (Salisachs et al., 1979; Harding и Thomas, 1980c; Westerberg et al., 1983).
Дифференцирование от типа 1 осуществляется при помощи электрофизиологического исследования. Моторная и сенсорная скорости проведения нормальны или только несильно снижены до 60% или больше от нормальных значений (Berciano et al., 1986). У пациентов с ШМТ старше двух лет моторная скорость проведения по п. medianus >38 м/с совместима с диагнозом синдром Шарко-Мари-Тута (ШМТ) 2, тогда как значения <38 м/с с большей вероятностью указывают на ШМТ 1 (Harding и Thomas, 1980с).
в) Гетерогенность синдрома Шарко-Мари-Тута (ШМТ) 2. ШМТ 2 связана, по крайней мере, с 10 локусами и 9 генами. В то время как ШМТ 2А была первоначально связана с миссенс-мутацией в гене из семейства кинезина 1B-ss (KIF1B) в одной родословной, в других пораженных семьях подобные мутации не идентифицировались. Недавно во многих таких семьях были обнаружены мутации в генетическом коде гибридного белка митохондриального слияния, митофузина 2 (MFN2), (Kijima et al., 2004; Zuchner et al., 2004).
Синдром Шарко-Мари-Тута (ШМТ) 2В вызывается миссенс-мутациями в маленьком эндосомальном белке ГТФ-азы RAB 7, который может играть свою роль в аксональном транспорте (Houlden et al., 2004). Она может быть перепутана с 1 типом НСВН из-за наличия умеренного сенсорного дефицита с осложнением в виде язв до 50% случаев (Verhoeven et al., 2003).
Для синдрома Шарко-Мари-Тута (ШМТ) 2С ген еще не найден, заболевание клинически характеризуется параличом диафрагмы, межреберных мышц и голосовых связок.
Синдром Шарко-Мари-Тута (ШМТ) 2D, вызывающая выраженную слабость рук, обычно с началом в подростковом возрасте, была локализована на участке хромосомы 7р15 и происходит из-за мутаций гена GARS, которые также могут вызвать синдром дистальной спинальной мышечной атрофии (СМА 5 типа).
Синдром Шарко-Мари-Тута (ШМТ) 2Е вызывается мутациями гена легкого белка нейрофиламента (NEFL). Скорости проведения по нерву могут перекрывать диапазоны демиелинизации и аксональной дегенерации. Случаи со сниженными скоростями проведения по нерву были классифицированы как относящиеся к ШМТ 1F, тогда как случаи со скоростями в диапазоне аксональных поражений были выделены в группу ШМТ 2Е. Случаи с замедленным проведением по нерву часто возникают до 13-летнего возраста, а при биопсии нерва выявляется смешанная картина аксонопатии/демиелинизации (Jordanova et al., 2003).
Несколько генов, обычно связываемых с аутосомно-доминантной демиелинизирующей невропатией, могут вызвать невропатию с аксональной дегенерацией. ШМТ 2I и 2J относятся к аксональным невропатиям, вызванным определенными мутациями гена MPZ, в то время как IIIMT2G и 2К вызваны, соответственно, доминантными и рецессивными повреждениями GDAP1.
Недавно были идентифицированы гены для двух дополнительных форм ШМТ 2 (L и F). Оба кодируют белки теплового шока, стрессовые белки, индуцируемые в ответ на множество физиологических факторов и факторов окружающей среды (Evgrafov et al., 2004; Tang et al., 2004, 2005).
Связь гломерулярной нефропатии с синдромом Шарко-Мари-Тута (ШМТ) 2 не представляется случайной при доминантном наследовании (Deniau et al., 1986). Накопление нейрофиламентов было найдено в одном типичном случае (Vogel et al., 1985).
г) Аутосомно-рецессивные формы синдрома Шарко-Мари-Тута (ШМТ) 2. Хотя известны аутосомно-рецессивные ШМТ аксонально-дегенеративных типов, немногие из них получили полное объяснение. Было показано, что мутации LMNA вызывают ШМТ 2В1 так же, как и множество других нарушений, включая аутосомно-доминантную мышечную дистрофию Эмери-Дрейфуса, конечностно-поясную мышечную дистрофию типа 1В и доминантную аксональную невропатию, связанную с мышечной дистрофией, заболеванием сердца и лейконихией (Goizet et al., 2004). LMNA — первый найденный ген, который вызывает и доминантную, и рецессивную ШМТ2. Некоторые мутации генов GDAP1 и MPZ изредка вызывают картину аксональной дегенерации.
Серии аксональных невропатий, описанные Ouvrier et al. (1981) и Gabreels-Festen et al. (1991), включали ряд аутосомно-рецессивных случаев с различным клиническим фенотипом, в то время как рецессивные случаи не были включены в классификацию наследственных невропатий. Начинающееся в первые пять лет жизни заболевание быстро прогрессировало, так что ко второй декаде большинство пациентов почти полностью парализовано ниже коленей и локтей. В пяти случаях не удалось измерить скорость проведения из-за отсуствия двигательной реакции, но у остальных пациентов они составляли более чем 35 м/с. У пяти из 10 пациентов из Сиднея в настоящее время удалось выявить мутацию генна митофузина 2 (mitofusin 2) (MFN2). Двое были смешанными гетерозиготами, то есть с рецессивным наследованием; трое имели доминантные (гетерозиготные) мутации.
Другой пациент из Сиднея с менее тяжелым фенотипом был гомозиготен по мутации с заменой фенилаланина на серин в 216 позиции в экзоне 7 гена MFN2 (неопубликованные личные наблюдения). Митохондрии проходят непрерывные циклы расщепления и слияния их внутренних и внешних мембран. MFN2 — это большая митохондриальная ГТФ-аза. Она может выполнять функцию фиксации митохондрии перед слиянием. Невропатия может таким образом произойти из-за дефекта в митохондриальном слиянии/делении, препятствующего выработке энергии и замедляющего аксональный транспорт. Много мутаций de novo.
Клиническая картина, вероятно, незначительно отличается от наблюдающейся при некоторых других рецессивных аксональных невропатях, где ген уже известен. Но об этом трудно судить по литературе, потому что зачастую во многих сериях клинические и патогистологические детали представлены в лучшем случае отрывочно.
Помимо аксональных дегенеративных форм ШТМ, перечисленных в таблице ниже, известно о других аутосомно-рецессивных аксональных полиневропатиях, которые по историческим или другим причинам традиционно не включались в классификации ШМТ. Они включают тяжелую инфантильную аксональную невропатию с нарушением дыхания, гигантоаксональную невропатию (см. ниже) и синдром Андерманн, невропатию, описанную у франкоканадцев, которая связана с агенезией corpus callosum, возникая, как было недавно выяснено, из-за мутаций гена SLC12A6, кодирующего белок ко-транспортера хлорида калия КСС3А (Casaubon et al., 1996; Howard et al., 2002).
д) Тяжелая инфантильная аксональная невропатия с нарушением дыхания. Другая, недавно охарактеризованная аутосомно-рецес-сивная аксональная невропатия, частично совпадающая со спинальной мышечной атрофией, была обозначена как тяжелая инфантильная аксональная невропатия с нарушением дыхания (severe infantile axonal neuropathy with respiratoryfailure — SIANR) (Wilmshurst et al., 2000) или спинальная мышечная атрофия с респираторным заболеванием (spinal muscular atrophy with respiratory disease — SMARD) (Grohmann etal., 2001) в зависимости от степени поражения периферических нервов. Это состояние описывается в отдельной статье на сайте.