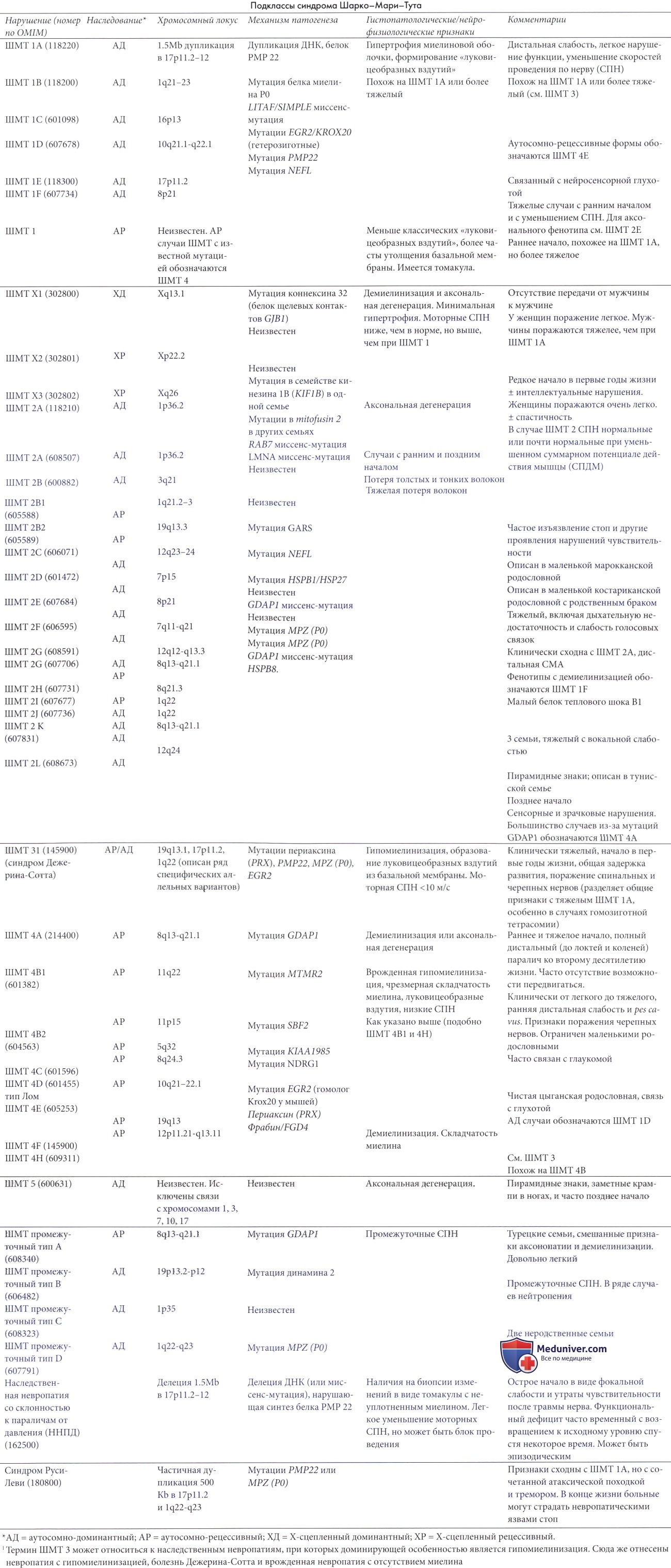
Аутосомно-рецессивные демиелинизирующие типы болезни Шарко-Мари-Тута (ШМТ 4, аутосомно-рецессивная НСМН типа 1)
Зафиксировано несколько пациентов с клинической картиной, похожей, но часто более тяжелой, чем при классической ШМТ 1 с аутосомно-рецессивным наследованием (Harding и Thomas 1980а; Gabreels-Festen et al., 1992; Gabreels-Festen и Gabreels, 1993).
Болезнь Шарко-Мари-Тута (ШМТ) 4 включает эту группу аутосомно-рецессивных демиелинизирующих невропатий. Это достаточно тяжелые заболевания с ранним началом и значительно сниженными моторными скоростями проведения (обычно около 20 м/с с диапазоном 3-37 м/с). У пораженных детей, вероятно, должны быть сходно пораженные сибсы при нормальных родителях, или родители, состоящие в кровном родстве. Недавние сообщения проливают свет на клинические и молекулярные находки при многих из этих заболеваний и обеспечивают некоторое понимание их относительной частоты.
В североафриканских и ближневосточных популяциях вероятной представляется более высокая распространенность.
Болезнь Шарко-Мари-Тута (ШМТ) 4А вызвана мутацией в гене ганглиозид-индуцированного связанного с дифференцировкой белка 1 (GDAP11 — ganglioside-induced differentiation-associated protein-1). Большинство пациентов демонстрирует фенотип с ранним началом и тяжелым течением с заметными деформациями стопы (Nebs et al., 2002). Клинический спектр этого заболевания был расширен за счет установления факта, что у пациентов с мутациями GDAP1 в дальнейшем может развиться паралич гортани и диафрагмы (Stojkovic et al., 2004).
Мутации GDAP1 могут вызвать демиелинизирующие или аксональные дегенеративные изменения в биоптатах нервов в различных семьях (Di Maria et al., 2004, Stojkovic et al., 2004).
Болезнь Шарко-Мари-Тута (ШМТ) 4В1 и ШМТ 4В2 характеризуются наличием в биопстате поражения миелина в складках и за их пределами, хотя эта находка не постоянна (Quattrone et al., 1996, Parman et al., 2004). Оба нарушения связаны с мутациями в генах белков из семейства миоту-буларина. Случаи болезни Шарко-Мари-Тута (ШМТ) 4В1 обычно начинаются рано, в основном с дистальной слабости, за которой следуют проксимальная слабость и атрофия. Глубокие сухожильные рефлексы обычно отсутствуют даже на ранних стадиях. Сенсорные изменения трудно выявить в грудном возрасте, но обычно они проявляются позже, в детстве.
В большинстве случаев имеется pes cavus. Имеются сообщения о птозе, внутренней и наружной офтальмоплегии. Слабость лицевого нерва может приводить к виду с «надутыми» губами. Позднее могут развиваться дисфагия, парез голосовых связок, дизартрия, атаксия и ночная гиповентиляция. Клиническое течение обычно тяжелее, чем при болезни Шарко-Мари-Тута (ШМТ) 1А. Нередко встречается сколиоз. Многие взрослые прикованы к инвалидному креслу, и несколько из них умерли в четвертой и пятой декадах жизни. Болезнь Шарко-Мари-Тута (ШМТ) 4В1 вызывается мутациями гена MTMR2.
Помимо патологических изменений миелина, болезни Шарко-Мари-Тута (ШМТ) 4В2 обычно связана с врожденной или ювенильной глаукомой, хотя в некоторых семьях, в которых причиной болезни была мутация MTMR13, этот симптом отсутствовал (Hirano et al., 2004).
Болезнь Шарко-Мари-Тута (ШМТ) 4С обычно проявляется в первые десять лет жизни с высокой частотой сколиоза. У некоторых пациентов развитие идет медленно, позволяя ходить вплоть до пятого десятилетия жизни, тогда как другие становятся зависящими от инвалидного кресла в подростковом возрасте (Senderek et al., 2003). Заболевание возникает из-за мутаций гена KIAA1985. Диапазон скоростей проведения по нерву от 4 до 37 м/с. Встречается заметное отличие в замедлении проведения между нервами одной и той же конечности (Gabreels-Festen и Thomas, 2005).
При гистопатологическом исследовании биоптата нерва обычно выявляются луковицеобразные вздутия базальной пластинки, вытянутые отростки шванновских клеток и случайные кольца, окружающие миелинизированные и немиелинизированные волокна. Также могут наблюдаться гигантские аксоны (Vallat et al., 2005).
Болезнь Шарко-Мари-Тута (ШМТ) 4D (HCMH-Lom) является наиболее частой из трех рецессивно наследуемых полиневропатий, которые в значительной степени ограничиваются популяциями этнических цыган. HCMH-Lom была первоначально идентифицирована у цыган, живущих в городе Ломе, Болгария. Она возникает из-за мутации гена NDRG1, который может кодировать в шванновской клетке сигнальный белок, необходимый для выживания аксона. Клиническая картина подобна другим аутосомно-рецессивным демиелинизирующим невропатиям, но обычно в возрасте после 10 лет возникает глухота (Kalaydjieva et al., 2000).
Болезнь Шарко-Мари-Тута (ШМТ) 4Е является редкой невропатией, связанной с гомозиготными мутациями гена EGR2 (ранняя реакция роста — early growth response), что может вызывать фенотип невропатии Дежерина-Сотта или врожденной гипомиелинизирующей невропатии (Warner et al., 1999). EGR2 регулирует экспрессию периаксина (periaxin). Скорости проведения по нерву очень низкие, часто меньше, чем 5 м/с.
У пациентов с болезнью Шарко-Мари-Тута (ШМТ) 4F, которая вызывается мутациями гена периаксина, обычно был клинический фенотип тяжелой ШМТ (Boerkoel et al., 2001; Guilbot et al., 2001). Возможно выраженное сенсорное поражение с дизэстезией и атаксией. Скорости проведения по нерву обычно очень низкие (2-3 м/с). Биопсии нерва показали тяжелую потерю миелинизированных волокон со многими луковицеобразными вздутиями и некоторые волокна с миелином за пределами складок. Мыши с недостатком периаксина демонстрируют сходные патогистологические признаки.
Болезнь Шарко-Мари-Тута (ШМТ) 4G (HCMH-Russe) является еще одной из невропатий, замеченных в популяции европейских цыган. Она похожа на ШМТ 4D, но без нейросенсорной глухоты. Она картирована на 10q22, но ген еще не идентифицирован (Thomas et al., 2001). ШМТ4Н очень похожа на болезнь Шарко-Мари-Тута (ШМТ) 4В1, но происходит из-за мутации гена FGD4, который кодирует фрабин, фактор обмена гуаниновых нуклеотидов. Фрабин может взаимодействовать с миотубуларином (см. ШМТ 4В1) (Stendel et al., 2007).
Другие формы болезни Шарко-Мари-Тута 4 очень редки или не полностью охарактеризованы в настоящее время.
Предполагалось, что болезнь Шарко-Мари-Тута (ШМТ) 4А может составлять до 25% случаев аутосомно-рецессивных ШМТ, но только одна мутация GDAP1 была обнаружена в недавнем исследовании 20 турецких пациентов с вероятной аутосомно-рецессивной демиелинизирующей невропатией с ранним началом, которые были тщательно изучены со скринингом на девять возможных этиологических факторов. Другие пять доказанных мутаций включают два случая мутации MTMR2, и по одному 1985 KIAA, NDRG1 и PRX. Таким образом, ни одна мутация не была обнаружена в 14 из этих 20 случаев (Parman et al., 2004). В подобном исследовании в Японии у 3 из 66 пациентов с болезнью Шарко-Мари-Тута (ШМТ) 4 были найдены мутации периаксина (Кapmа et al., 2004).