Причины и классификация болезни Шарко-Мари-Тута 1 (ШМТ, наследственной моторной и сенсорной невропатии 1 типа, НМСН)
Наследственные моторные и сенсорные невропатии (НМСН) являются наиболее распространенными дегенеративными поражениями периферической нервной системы, составляя приблизительно 40% хронических невропатий детского возраста (Ouvrier, 1992). Дегенерация миелиновых оболочек и/или аксонов вызывает преимущественно дистальную паралитическую амиотрофию, в основном поражающую нижние конечности и сопровождающуюся арефлексией. Моторное поражение затеняет сенсорные нарушения.
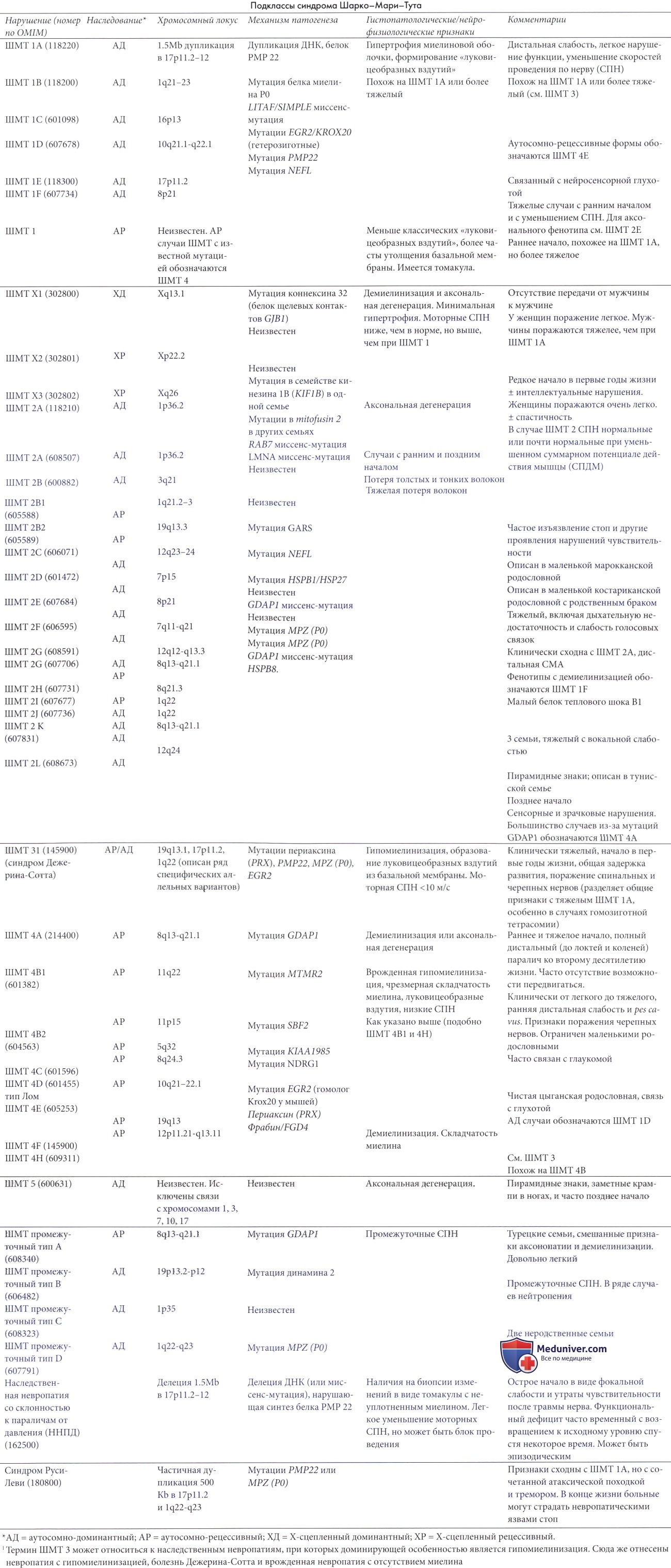
В основе действующей классификации наследственных моторных и сенсорных невропатий (НМСН) сохранились патоморфологические элементы (аксональная или демиелинизирующая), клинико-электрофизиологических проявлений (особенно моторная и сенсорная скорость проведения) и тип наследования. Пporpecc в молекулярной генетике быстро меняет критерии классификации. Многие из НМСН заканчиваются клинической картиной, называемой болезнь Шарко-Мари-Тута (ШМТ). Не так давно неврологи чаще использовали термин НМСН, тогда как генетики предпочитают ШМТ. Тип 1 НМСН синонимичен ШМТ 1, но есть некоторые несоответствия в номенклатуре, особенно в отношении более редких форм.
Например, ШМТ4 классифицирована как демиелинизирующая форма аутосомно-рецессивной НСМН, тогда как НСМНIV типа исходно соотносится с болезнью Рефсума. В данной главе будет использоваться преимущественно номенклатура ШМТ. В таблице ниже представлена комбинированная классификация.
Для патологических изменений при болезни Шарко-Мари-Тута (ШМТ) 1 характерна обширная сегментарная демиелинизация и ремиелинизация с развитием луковицеобразных вздутий вокруг нервных волокон, снижением моторных и сенсорных скоростей проведения по нервам и доминантным наследованием. Тем не менее, есть сообщения как об аутосомно-рецессивных (Harding и Thomas, 1980а; Gabreels-Festen et al., 1992), так и Х-сцепленных формах (Hahn et al., 1990, Ionasescu et al., 1991). Распространенность НМСН I 3,8 на 100000 населения, что составляет приблизительно 50% педиатрических случаев наследственных невропатий. Хотя пенетрантность болезни высока (83%), имеет место значительная вариабельность клинической и патоморфологической выраженности (Harding и Thomas, 1980b).
В некоторых случаях клинические и даже морфологические (биопсия нерва) данные иногда минимальны, особенно у маленьких детей. Тем не менее дети, пораженные ШМТ 1 А, почти всегда демонстрировали некоторые симптомы, достигая возраста трех лет. Встречаются умеренные и даже бессимптомные случаи (Thomas et al., 1997), и поэтому систематическое обследование родителей и родственников улучшает диагностические возможности при предварительном консультировании. Большая часть дупликаций de novo имеет отцовское происхождение (Palau et al., 1993; Harding, 1995).
Болезнь Шарко-Мари-Тута (ШМТ) 1 является генетически гетерогенным типом, но более 80% случаев в детстве связаны с хромосомой 17 (ШМТ1А). Обычно дефект в таких случаях — субмикроскопическая дупликация, длиной приблизительно 1,5 мегабаз, в пределах участка 17р11.2 (Raeymaekers et al., 1991). Дупликация включает ген, кодирующий белок периферического миелина 22 (peripheral myelin protein 22 — РМР22) (Patel et al., 1992) так, чтобы ген был представлен в трех копиях. В нескольких случаях без дупликации была найдена точечная мутация гена РМР22 (Roa et al, 1993), что указывает на то, что этот ген — ген болезни при ШМТ 1.
Предполагается, что дупликация является следствием неравного кроссинговера во время мейоза. Делеция того же участка приводит к наследственной невропатии со склонностью к параличам от сдавления нерва (ННПД), описанной ниже. Также были зарегистрированы случаи ШМТ 1А из-за трисомии 17. Непонятен механизм, за счет которого чрезмерная экспрессия РМР22 (вызывемая или наличием трех копий гена РМР22 или приобретением функциональной мутации) приводит к клиническим проявлениям. Модели на мышах и крысах с повышенной экспрессией РМР22 развивают фенотипические признаки, согласующиеся с ШМТ 1, и тяжесть демиелинизации пропорциональна уровню экспрессии РМР22. РМР22 содержит лишь 2-5% миелина периферической нервной системы. При некоторых связанных с РМР22 невропатиях вероятным механизмом представляется нарушение клеточного транспорта (Lupski и Chance, 2005).
В отдельных семьях болезнь вызывается мутациями в гене MPZ (myelin Р0) (ШМТ 1В), который картирован на участке хромосомы 1q21-q23 (Warner et al., 1996). Мутации генов, как РМР22, так и MPZ, вызывают разнообразные клинические картины, варьирующие в зависимости от локализации и тяжести мутации от врожденной гипомиелинизации, через фенотип Дежерина-Сотта к фенотипу ШМТ 1 или даже, в случае MPZ, до аксонального дегенеративного синдрома ШМТ (ШМТ2) (Warner et al., 1996). По нашему опыту педиатрических наблюдений, клинические проявления случаев, вызванных точечными мутациями, тяжелее, чем при типичных случаях ШМТ 1 А, связанных с дупликацией.
Результаты патогистологического исследования ШМТ 1В подобны таковым при болезни Шарко-Мари-Тута (ШМТ) 1 А, но в значительном количестве волокон может быть замечен некомпактизированный миелин, и очаговая складчатость миелина («колбаски») может также быть значительной. MPZ составляет приблизительно 50% белка миелина и важен для адгезии миелина (Shy, 2005).
Некоторые случаи не картированы ни в одном из этих локусов. В некоторых доминантно наследуемых случаях, соотносимых с болезнью Шарко-Мари-Тута (ШМТ) 1C, выявлялись мутации гена LITAF/SIMPLE, который может быть вовлечен в аномальную деградацию белка (Street et al., 2003).
Смущает, что болезнь Шарко-Мари-Тута (ШМТ) 1D относят к ШМТ 1, обусловленной гетерозиготной мутацией, вызывающей дефект «цинковых пальцев» регулятора транскрипции EGR2 (также известного как Кгох20), который индуцирует транскрипцию многих генов, важных для формирования и поддержания уровня миелина (Warner et al., 1999). Гомозиготные EGR2 мутации вызывают более тяжелую аутосомно-рецессивную гипо- и демиелинизирующую невропатию ШМТ 4Е.
Х-сцепленная болезни Шарко-Мари-Тута (ШМТ X). Приблизительно в 10% случаев заболевание сцеплено с Х-хромосомой, где было выявлено по меньшей мере три локуса (Ionasescu et al., 1991, 1992). Самая частая (Х-сцепленная доминантная) форма находится на длинном плече на Xq13.1. Известно, что многочисленные мутации в этом очаге вовлекают ген коннексина 32 (Bergoffen et al., 1993), связывающего мембраны в зоне щелевых контактов белка, примыкающего к перехватам Ранвье и насечки Шмидта-Лантермана. Изучение родословных показывает ожидаемое отсутствие передачи от мужчины к мужчине. Обычно выраженность поражения у мужчин значительно выше, чем у женщин, но в целом клинические поражения в первое десятилетие менее тяжелы, чем при ШМТ 1 в том же возрасте. С другой стороны, возможная степень инвалидизации у взрослых мужчин больше при ШМТ X, чем при ШМТ 1.
В некоторых случаях у пораженных детей наблюдается изолированная глухота (Stojkovic et al., 1999), а у некоторых из них отмечались преходящие симптомы со стороны ЦНС, связанные с поражением белого вещества (Haneman et al., 2003). Нейрофизиологические признаки Х-сцепленного типа у мужчин могут напоминать таковые при ШМТ 1, но средняя скорость проведения по нерву (СПН) приблизительно на 10 м/с больше у мужчин с ШМТ Х (средняя перонеальная СПН 31 м/с), чем у мужчин с ШМТ 1 (средняя перонеальная СПН 22 м/с) (Nicholson и Nash, 1993), тогда как у женщин она ближе к аксональному типу. Представляется, что большинство «промежуточных» типов ШМТ с умеренно сниженными скоростями проведения являются Х-сцепленными. Существуют и другие варианты, к примеру, мутации допамина, являющиеся типичной причиной промежуточной формы ШТМ 1, иногда с нейтропенией (Zuchner et al., 2005).
Редкая Х-связанная рецессивная аксональная форма картирована на Хр22.2, и другая аксональная форма, связанная с глухотой и задержкой интеллектуального развития, картирована на Xq24-q26 (Priest et al., 1995; Ouvrier et al., 2007).