Синдром Ретта является необычным нарушением развития мозга, с напоминающими аутизм или нейродегенеративное заболевание признаками, но в действительности не является ни одним из них. Синдром Ретта представляет собой характерный комплекс клинических проявлений, включающих раннюю психомоторную регрессию с аутистическими проявлениями, замещение целенаправленных действий рук стереотипными движениями, атаксией и апраксией при ходьбе и приобретенной микроцефалией (Hagberg, 1989). В таблице 4.4 представлены международные критерии диагностики (Rett Syndrome Diagnostic Criteria Work Group, 1988; Trevathan и Naidu, 1988). За редким исключением данное заболевание встречается только у девочек (Zoghbi, 1988, Hagberg, 1989).
а) Распространенность. Распространенность синдрома Ретта в Швеции и западной Шотландии, составляет 1 на 10000 и 1 на 18000 девочек (Kerr и Stephenson, 1985; Hagberg, 1993; Bienvenu et al., 2006).
б) Патогенез. Синдром Ретта приблизительно в 80% случаев связан с мутациями гена МЕСР2, и длительное время считалось, что мутация данного гена является причиной синдрома Ретта. Тем не менее, фенотипические проявления мутаций гена МЕСР2 разнообразны и включают задержку умственного развития с припадками или без них, фенотип, подобный синдрому Ангельмана, и аутизм (Zoghbi, 2005). Как минимум еще один ген (CDKL5) связан с развитием судорожного варианта заболевания. Ген МЕСР2 является ингибитором фактора транскрипции, способным отключать несколько важных для развития головного мозга генов, а в связи с экспрессированием в различных типах клеток и органов— влиять на соматическое развитие в целом. В этой связи, представляется вероятным, что мутация гена МЕСР2 при синдроме Ретта является частью последовательной цепи событий, приводящих к развитию ряда сцепленных с Х-хромосомой нарушений развития нервной системы.
В настоящее время синдром Ретта представляется скорее патологией развития, а не дегенеративным заболеванием (Naidu, 1997). Структурные аномалии мозга выражены слабо и включают маленький размер мозга с плотно расположенными нейронами и снижением клеточных процессов. В подавляющем большинстве случаев заболевание не имеет наследственного характера, несмотря на то, что выборочное поражение девочек предполагает генетическое происхождение синдрома.
в) Клинические проявления. Течение синдрома Ретта имеет необычный характер. Заболевание начинается как прогрессирующее состояние с более или менее стремительной деградацией с утратой ранее полученных навыков.
Течение заболевания может быть разделено на четыре стадии. Дебют клинических симптомов приходится на возраст от шести месяцев до трех лет, в большинстве случаев заболевание манифестирует до 18 месяцев. Изначально развитие ребенка может не отличаться от нормы, но у больных девочек часто с рождения отмечается гипотония и слегка замедленное развитие (Einspieler et al., 2005).
Большинство девочек с синдромом Ретта развиваются нормально или почти нормально до 6-16 месяцев. Ретроспективно часто отмечается умеренная гипотония и минимальная задержка развития. Освоение целенаправленных движений рук является предварительным условием постановки диагноза. В дальнейшем у многих пациентов отмечается остановка развития или резкая утрата навыков (возможна потеря социальной улыбки, способности к взаимодействию и некоторых речевых навыков). Основным проявлением является утрата мануальных навыков. Данное проявление часто развивается стремительно в течение нескольких недель или носит «взрывной» характер, возникая в течение нескольких дней. Некоторые, но не все, дети становятся отчужденными, эмоционально отстраненными и описываются как «аутисты». У других медленно развивается малоэмоциональный стиль социального взаимодействия, иногда определяемый как аутистический.
Небольшое количество пациентов страдает от приступов ярости, тревоги, смущения и беспорядочной гиперактивности. При наличии аутистической или подобной аутистической фазы данное состояние может длиться от одного месяца до нескольких лет. Обычно к достижению школьного возраста (или не позднее пубертатного периода) аутистические симптомы начинают убывать, но не во всех случаях. По имеющимся данным, у большинства аутистов, вне зависимости от причины заболевания, присутствует одинаковый характерный тип развития.
Для девочек с синдромом Ретта характерны различные виды стереотипных движений рук, большая часть которых включают «движения в области средней линии», то есть обе руки «моются» или складываются по средней линии, обе руки засовываются в рот или шлепают по средней линии лба или шеи. На ранней стадии возможны более типичные для аутизма стереотипии с хлопаньем в ладоши.
У некоторых больных девочек зарегистрированы случаи смеха в середине ночи. Данное проявление возникает в результате нейрометаболических/неврологических нарушений в головном мозге (например, при мукополисахаридозе Санфилиппо) и встречается у многих девочек с аутизмом, не страдающих синдромом Ретта.
Бруксизм и гипервентиляция являются типичными проявлениями синдрома Ретта и иногда интерпретируются как признаки чрезвычайной тревожности, что не подтверждается опытом.
Третья стадия заболевания характеризуется медленным появлением неврологических признаков, таких как пирамидные знаки. Эпилептические припадки на данной стадии развиваются у 2/3-3/4 больных. Нередки предшествующие изменения ЭЭГ, включающие ритмичную тета-активность в лобно-центральной области, пароксизмальные проявления (пики или комплексы «пик-волна»), часто локализованные в задних отделах, вспышки медленных комплексов «пик-волна» (особенно во время сна) и прогрессирующее замедление и деградация фонового ритма (Niedermeyer et al., 1986, Glaze et al, 1987).
Частым проявлением является гипервентиляция с дыхательными паузами, иногда вызывающая синкопальные состояния, которые могут быть ошибочно приняты за эпилептические припадки. От двух третей до трех четвертей пациентов не способны к самостоятельному передвижению, а с прогрессированием четвертой стадии в большинстве случаев эта способность окончательно утрачивается (Hagberg, 1989). Патология роста встречается в большинстве случаев. Чрезвычайно часто встречающийся кифосколиоз является одним из основных осложнений синдрома. Устойчивый лабораторный маркер не выявлен. Первично зарегистрированная гипоаммониемия обнаруживается только в редких случаях и обычно обусловлена внешними факторами, например, лечением вальпроатами.
На МРТ выявляется уменьшение объема мозга, преимущественно за счет белого вещества, уменьшение объема хвостатого ядра и среднего мозга и нормальное строение извилин (Reiss et al., 1993).
г) Разновидности синдрома Ретта. Фенотип девочек с синдромом Ретта отличается большим разнообразием и варьирует от врожденных форм с практически полным отсутствием психического развития до легких форм, при которых может сохраняться способность к передвижению и даже речь (Huppke et al., 2003). Выделяют «скрытые формы», при которых проявления заболевания типичны, но выражены слабо; тяжелые врожденные формы, при которых практически полностью отсутствует психическое развитие; умеренные формы с регрессом в позднем детском возрасте; скрытые формы с широким диапазоном проявлений и варианты заболевания с сохранением речи (Hagberg и Skjeldal, 1994). В случае сохранения речи девочки могут обладать словарным запасом от 20 до нескольких сотен слов, а некоторые говорят длинными (обычно в виде эхолалий) предложениями (Zappella et al., 1998, 2001).
Также описана форма заболевания с проявлениями синдрома Ангельмана (Hitchins et al., 2004). Судорожный вариант заболевания характеризуется ранним началом припадков в виде инфантильных спазмов, которые могут предшествовать другим типам тонических или фокальных припадков и резистентны к лечению. Данная форма заболевания связана с мутациями гена CDKL5 (Weaving et al., 2004). Тем не менее, мутация данного гена выявляется не во всех случаях; вероятно, не все случаи мутации гена CDKL5 проявляются синдромом Ретта. Синдром Ретта был выявлен только у одного из семи пациентов с данной мутацией, у которых отмечались инфантильные спазмы и выраженная задержка умственного развития (Archer et al., 2006). Зарегистрированы редкие случаи типичного синдрома Ретта у мальчиков (Matsuyama et al., 2005). Среди мальчиков-носителей мутации чаще отмечается энцефалопатия новорожденных без типичных для синдрома проявлений (Moog et al., 2006).
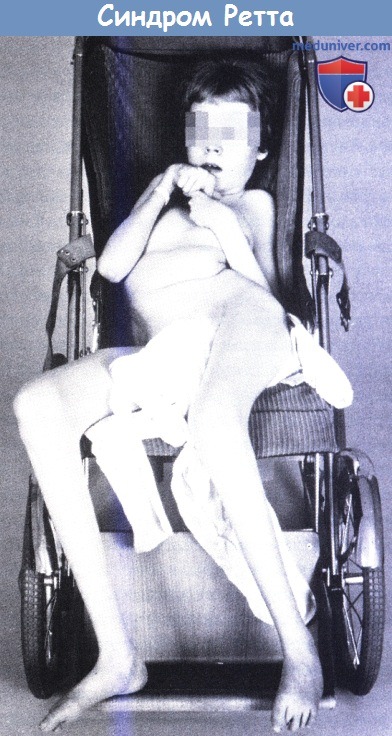
Синдром Ретта у 15-летней девочки.
Характерные стереотипные движения кистей, выраженный сколиоз и атрофия нижних конечностей.
д) Диагностика. Диагноз синдрома Ретта до сих пор устанавливается на основании анамнеза и клинических проявлений, а обнаружение мутации гена МЕСР2 во многих случаях служит подтверждением (Huppke и Gartner, 2005).
Диагностические критерии синдрома Ретта приведены в таблице ниже. У многих девочек с синдромом Ретта на ранних стадиях заболевания отмечаются аутистические проявления без четко выраженных неврологических отклонений. Поэтому в возрасте до трех лет часто ставится диагноз аутизма. Диагноз синдрома Ретта учитывается во всех случаях выявления симптомов аутизма у девочек в очень раннем детском возрасте. В исследовании Witt-Engerstrom и Gillberg (1987) девочек с синдромом Ретта в 80% случаев изначально предполагался аутизм или проявления аутизма, но на основе имеющихся данных о распространенности было установлено, что в 1/3-1/2 случаев выявления симптомов аутизма в течение первых лет жизни в итоге определялся симптом Ретта.
В редких случаях синдром Ретта характеризуется необычно медленным течением. В таких случаях диагноз «чистого аутизма» с серьезной/умеренной умственной отсталостью может не меняться в течение многих лет. Частая встречаемость атипичных форм (Goutieres и Aicardi 1985), отсутствие надежного маркера во многих случаях должны предостеречь от окончательного диагноза. Возможность выявления синдрома Ретта и задержки умственного развития у сибсов позволяет предположить наличие широкого спектра проявлений, но для подтверждения диагноза необходим определенный тест (Huppke et al., 2003). Hagberg и Skjeldal (1994) представили предварительные диагностические критерии для разновидностей синдрома Ретта.
е) Дифференциальная диагностика синдрома Ретта. В основном необходима дифференциальная диагностика с синдромом аутизма. Нейронный восковидный липофусциноз новорожденных с движениями рук, имитирующими вязание, описанный Santavuori, может иметь сходство с синдромом Ретта, но редко встречается за пределами Финляндии (Santavuori et al., 1973; Santavuori 1988). Дефицит орнитинтранскарбомилазы также ошибочно может приниматься за синдром Ретта. Синдром Ангельмана может иметь сходство с синдромом Ретта в связи с судорожной атаксией, наблюдаемой в обоих случаях помимо проявлений аутизма, задержки умственного развития и припадков. Исследование хромосом в сложных случаях позволяет дифференцировать заболевания.
Среди случаев с пограничными симптомами синдрома Ретта и аутизма (Gillberg, 1989), включая «скрытые формы» и формы заболевания с сохранением речи (Hagberg и Rasmussen, 1986), часто отмечаются многие классические проявления синдрома Ретта, но они не соответствуют всем критериям; обычно они также соответствуют большинству или всем критериям аутистического расстройства (или детского аутизма). У некоторых девочек классические симптомы аутизма проявляются только после длительного преморбидного периода (или 1 стадии) заболевания. Ретт-подобные симптомы встречаются также в сочетании с другими неврологическими расстройствами, такими как синдром Мебиуса (Gillberg и Steffenburg, 1989) и мукополисахаридоз.
ж) Лечение. Лечение синдрома Ретта неэффективно. При попытке применения бромокриптина и налоксона положительных результатов получено не было. Пациентам необходима физиотерапия и внимательное отношение к деталям обыденной жизни.
Стимуляторы показаны девочкам с хорошей реакцией на лечение в течение раннего периода отмены препарата. Важной частью терапии синдрома Ретта является ортопедическое лечение для предупреждения развития или уменьшения выраженности сколиоза.
Лечение поведенческих/психиатрических отклонений, вызванных синдромом Ретта, требует знания естественного течения заболевания, чтобы такие симптомы как аутизм и ночной смех не интерпретировались ошибочно как специфические психологические отклонения или проблемы общения. Восприятие речи при синдроме Ретта чрезвычайно снижено. Общение осуществляется с помощью зрительного контакта и жестов. Некоторые функции рук могут быть восстановлены при длительной ежедневной тренировке каждой руки в отдельности.
При применении бромокриптина (20 мг/кг в сутки) были достигнуты некоторые положительные результаты (Zappella et al., 1990), но для подтверждения эффективности необходимо проведение двойных слепых пла-цебо-контролируемых исследований. Результаты применения налоксона неоднозначны.
з) Исход. Отдаленные исходы синдрома Ретта известны только частично. Продолжительность жизни относительно увеличена, некоторые пациенты достигают возраста 80 лет и более. Подавляющее большинство (практически все) пациенты имеют чрезвычайно выраженные неврологические и/или умственные отклонения и зависят от других людей практически во всех областях повседневной жизни. В большинстве случаев клиническая картина осложняется эпилепсией, запорами, сколиозом, прогрессирующими двигательными (и вазомоторными) отклонениями. Психиатрические/по-веденческие отклонения могут являться предметом озабоченности в детском и иногда в подростковом возрасте, но обычно они в меньшей степени затрагивают пациентов старшей возрастной группы.