а) Генетические причины сенсоневральных нарушений слуха у ребенка. Это быстро развивающаяся область исследований с традционным разделением на синдромальные (30%) и несиндромальные (70%) причины (Friedman и Griffith, 2003; Nance, 2003). Подобное подразделение удобно при первичном клиническом диагнозе, но следует учитывать, что некоторые синдромальные состояния могут легко оказаться несиндромальными, так как более широкие проявления болезни не были выявлены, например, удлиненные интервалы QT при синдроме Ервелла-Ланге-Нильсена, дисфункция щитовидной железы при синдроме Пендреда, и пигментный ретинит при синдроме Ашера. Различия на молекулярном уровне также могут вводить в заблуждение.
Хотя 50% несиндромальных случаев сенсоневральной потери слуха вызывается мутациями коннексина-26, другие гены коннексина могут иметь сопутствующие аномалии, например, дерматологические нарушения при изменениях коннексина-30 и коннексина-31, пальмо-плантарный гиперкератоз с доминантным G59A аллелем, и неврологические аномалии при патологии коннексина-32, наблюдаемые при Х-сцепленной болезни Шарко-Мари-Тута.
Около 33 рецессивных, 41 доминантного и пяти Х-сцепленных локусов были картированы при несиндромальной генетической слепоте. Несмотря на очень большое число идентифицированных локусов, большинство случаев генетической слепоты вызваны мутациями, поражающими один ген из коннексинов (Denoyelle et al., 1999).
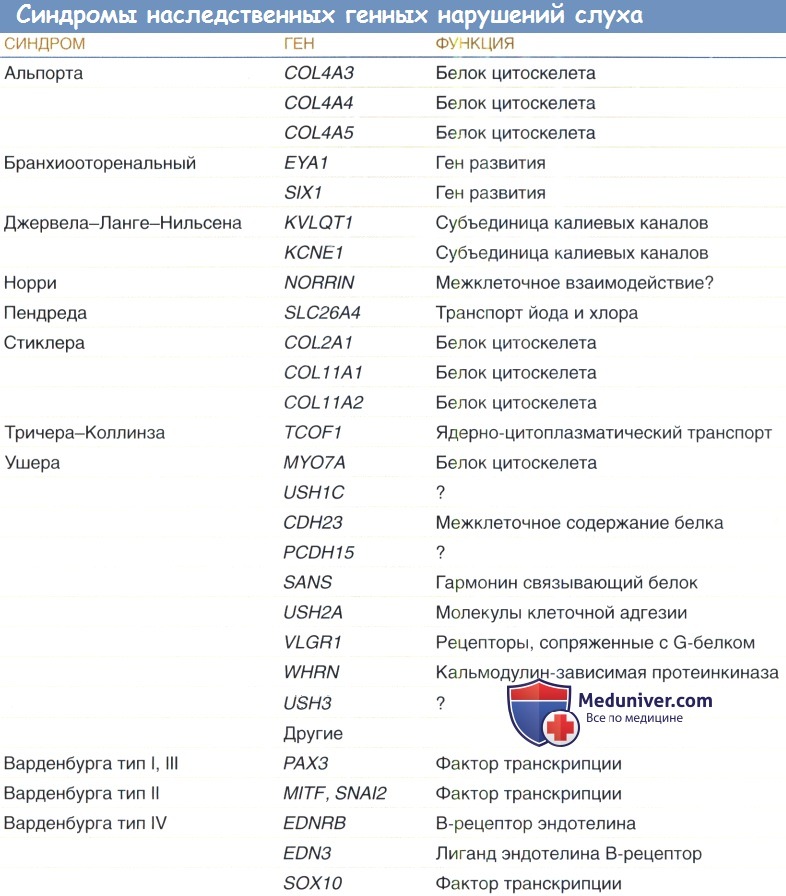
Коннексины — семейство генов, кодирующих субъединицы белков щелевидных соединений. Щелевидное соединение образуется при соединении двух гексамеров геми-коннексинов соприкасающихся клеток. По образовавшемуся каналу происходит ток ионов и небольших молекул между клетками. Определение гена GJB2 в локусе 13q12— 13 — один из наиболее часто применяемых в клинике тестов для диагностики несиндромальной глухоты, также применяются тесты на SLC26A4 и WFS1 (Smith, 2004). Эти гены определяют значительное число случаев в общем числе мутаций, вызывающих генетическую глухоту, скрининговые исследования для их выявления относительно просты.
GJB2 в локусе DFNB1 вызывает аутосомно-рецессивную несиндромальную глухоту. GJB2 кодирует трансмембранный белок коннексин-26, который вместе с пятью другими коннексинами составляет коннексон, являющийся структурным компонентом щелевидного соединения. Тяжесть потери слуха при мутации гена коннексина-26 крайне вариабельна и не поддается прогнозированию даже внутри одной семьи, хотя при наблюдении молодых людей было продемонстрировано, что в большинстве случаев потеря слуха не прогрессирует (Denoyelle et al, 1999).
Ген DFNB1, вызывающий эту рецессивную несиндромальную сенсоневральную потерю слуха, вызывает примерно 15% всех случаев потери слуха у младенцев. Выполнение теста на эту часто встречающуюся мутацию имеет большое значение, так как она встречается в 95% случаев в семьях европеоидной расы не от родственных браков. Если тест положительный, необходимость проведения КТ, пробы с перхлоратом, или поиска пигментного ретинита отпадает(Объединенный комитет по проблемам слуха у младенцев, 2000).
Потеря слуха, вызванная мутацией DFNA36, является рано начинающимся быстро прогрессирующим состоянием доминантной несиндромальной глухоты (Makishima et al., 2004).
Мутации нескольких генов, кодирующих различные миоцины, психоскелетные двигательные белки, создающие напряжение или способствующие движениям компонентов клетки вдоль филаментов актина, связаны с потерей слуха: MY06, MY07A, MY015A, MYOIA и MY03A (Ben-Yosef и Friedman, 2003).
Митохондриальная мутация A1555G обусловливает повышенную чувствительность к нормальным дозам аминогликозидов, является примером ситуации, когда генетическая предрасположенность в комбинации с внешними факторами риска приводит к сенсоневральной потере слуха.
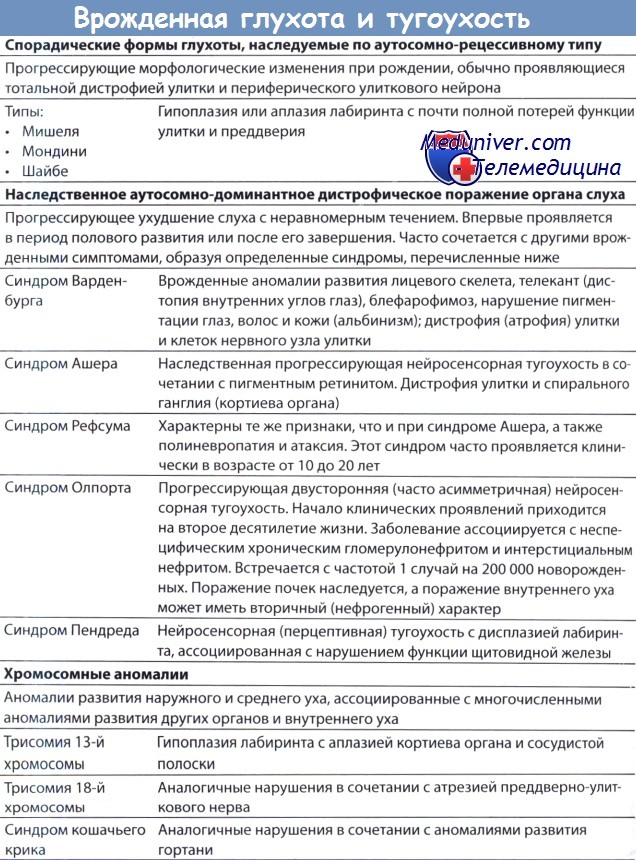
Генетические синдромы. Повторимся, прогресс в этой области идет очень быстро, и молекулярный анализ может подтвердить диагноз при все большем числе состояний: синдром Ваарденбурга I-IV типов, синдром Трешера Коллинза, синдром Альпорта, бранхио-оторенальный синдром, синдром Ашера, синдром Ервелла-Ланге-Нильсена, болезнь Шарко-Мари-Тута, и краниосиностоз-глухота. Три синдрома — Ервелла-Ланге-Нильсена, Пендреда и Ашера—вначале могут быть несиндромальными, пока не будет диагностировано поражение других органов.
1. Синдром удлиненного QT/синдром Ервелла-Ланге-Нильсена. Мутации различных генов калиевых каналов могут привести к синдрому длинного QT; если эти изменения сопровождаются врожденной сенсоневральной глухотой, такое состояние известно как синдром Ервелла-Ланге-Нильсена (ЕЛНС) (Neyroud et al, 1997). Распространенность у детей с врожденной глухотой составляет 0,21% (Ocal et al., 1997). Удлинение интервала QT наблюдается на ЭЭГ и отражает дефект реполяризации сердечной мышцы, который может привести к рецидивирующим синкопальным состояниям, желудочковой аритмии и внезапной смерти. Синкопальные состояния могут провоцироваться испугом.
Синдром длинного QT определяется на основании удлинения интервала QTc (корректированный QT) >470 мс у бессимптомных пациентов. Ген KVLQT1, вызывающий ЕЛНС, также вызывает синдром Романо Уорда, при котором не происходит потери слуха. KVLQT1 картирован на хромосоме 11р15.5 (локус JLNS1); в результате рецессивной мутации, поражающей другой ген калиевых каналов KCNE1 в локусе JLNS2 хромосомы 21q22.1, может развиваться идентичный фенотип с поражением системы активного транспорта калия в волосковых клетках улитки. Другой ген калиевых каналов, KCNQ4, ограниченный наружными волосковыми клетками, имеет мутации, вызывающие относительно частую доминантную форму прогрессирующей потери слуха с типичным началом в первые две декады жизни, первоначально поражающую восприятие высоких частот, и затем в течение десятилетия прогрессирующую до глубокой глухоты.
Всем детям с нарушением слуха должно выполняться ЭКГ, и, при получении пограничных или патологических результатов, ребенок должен направляться к кардиологу для дальнейшего обследования, так как могут потребоваться ЭКГ-мониторирование с проведением нагрузочных проб для выявления удлиненного интервала QT. Это связано с высоким риском развития синкопальных состояний и внезапной смерти в течение первого года жизни или позже; также наблюдается вестибулярная арефлексия.
2. Синдром Пендреда. Синдром Пендреда, возникающий в результате мутации в SLC26A4, является наиболее частой причиной синдромальной глухоты, вызывает более 5% случаев аутосомно-рецессивной потери слуха. Он характеризуется двусторонней сенсоневральной глухотой, которой сопутствует зоб, с гипотиреозом или без него (Reardon et al., 1997). Как оказалось, существует взаимосвязь между тяжестью нарушений слуха и степенью гипотиреоза. Глухота врожденная и сопровождается аномалиями височной кости, варьирующих от изолированного увеличения вестибулярного водопровода до дисплазии Мондини, более обширной мальформации, включающей также гипоплазию улитки (Napiontec et al., 2004). Потеря слуха обычно глубокая (хотя в начале заболевания степень ее выраженности различна), быстро прогрессирующая и односторонняя.
Зоб обычно развивается в возрасте старше 10 лет, дисфункция щитовидной железы вариабельна. Сенсоневральной глухоте иногда сопутствует нарушение вестибулярной функции.
3. Синдром Ашера. Синдром Ашера вызывает 2-4% всех случаев глубокой глухоты и 50% случаев глухоты со слепотой. Синдром Ашера I типа характеризуется вестибулярной дисфункцией, глубокой врожденной глухотой и рано развивающимся пигментным ретинитом. Прогрессирующая дегенерация сетчатки вызывает потерю ночного зрения и сужение полей зрения, приводящее к слепоте. I тип отличается от II типа ранним дебютом пигментного ретинита. При II типе также обычно бывает менее тяжелая потеря слуха и нормальная вестибулярная функция. Синдром Ашера III типа характеризуется прогрессирующей постлингвальной потерей слуха и пигментным ретинитом различной степени тяжести, отсутствием вестибулярного компонента. Синдром Ашера является фенотипически и генотипически сложным, и очевидно связан с локусом USH1, картированном на 10q21—22 и 11q13.5 (Ben-Yosef и Friedeman, 2003).
4. Синдром Альпорта. При этом состоянии потеря слуха может не проявляться до достижения возраста 8-10 лет, и у 50% больных развивается прогрессирующая двусторонняя потеря слуха, первоначально нарушающая восприятие высоких частот. Развивается прогрессирующий нефрит. Частота составляет 1 на 10000 новорожденных, встречаются Х-сцепленные, аутосомно-рецессивные и аутосомно-доминантные варианты. В первом десятилетии жизни наблюдается гематурия, диагноз ставится на основании гематурии или почечной недостаточности, характерных результатах биопсии почек, сенсоневральной потере слуха, и офтальмологических симптомах — переднем лентиконусе или отложениях в макулярной области. Глазная патология также может включать врожденные катаракты и сферофакию. Заболевание развивается вследствие мутаций, поражающих так или иначе одну из трех тканеспецифичных полипептидных субъединиц коллагена, кодируемых генами COL4A3, СО-L4A4 и COL4A5.
Ген COL4A5 локализован на Xq22, тогда как гены, кодирующие коллаген 3 и 4 расположены рядом на 2q35.
5. Синдром Альстрема. Синдром Альстрема — генетическое заболевание с рецессивным механизмом наследования, вызываемое мутацией гена ALMS1 (Hearn et al., 2002), которое характеризуется врожденной дистрофией сетчатки, приводящей к слепоте, сенсоневральному поражению слуха, ожирением в детском возрасте, инсулинрезистентным сахарным диабетом 2 типа и гипертриглицеридемией, которая может спровоцировать панкреатит и легочные симптомы (Michaud et al., 1996). Из 182 случаев, изученных Marshall et al. (2005), у 20% была отмечена неврологическая симптоматика, включая клонический тик и абсансные припадки, тогда как задержка двигательного или речевого развития наблюдалась почти в 50% случаев. Отмечались фиброзные изменения многих органов, включая почки, сердце, печень, легкие, мочевой пузырь, гонады и поджелудочную железу.
6. Синдром Ваарденбурга. Синдром Ваарденбурга поражает 1-2% больных с глубокой потерей слуха, которая может быть двусторонней или односторонней, и связана с дефектами структур и тканей — производных нервного гребешка (Liu et al., 1995). Пигментные нарушения включают ярко голубые глаза, полную или сегментарную гетерохромию, и участки гипер- или гипопигментации кожи. Отмечается смещение внутренней спайки век кнаружи, «нос попугая» и сросшиеся брови. Могут развиваться симптомы поражения желудочно-кишечного тракта, в анамнезе может отмечаться болезнь Гиршпрунга. С повышенной частотой отмечаются дефекты нервной трубки и конечностей. Существует, по крайней мере, восемь локусов, участвующих в формировании фенотипа. Синдром Ваарденбурга I типа может быть результатом более 50 различных мутаций, поражающих ген РАХЗ в 2q35.
Синдром Ваарденбурга ПА типа развивается при мутации в локусе MITF в 3q12, при этом состоянии аномалии век отмечаются реже, но более выражены глухота и гетерохромия. У некоторых пациентов с синдромом Ваарденбурга НА типа наблюдается альбинизм, с веснушчатостью или без, и это состояние известно как синдром Титце. Были описаны некоторые другие локусы, такие как при синдроме Ваарденбурга III типа, состояния, проявляющегося различными дефектами конечностей. Механизм, как оказывается, заключается в том, что поражаются клетки нервного гребешка улитки, участвующие в формировании сосудистой полоски, и так как в таком состоянии полоска не может поддерживать необходимый для нормального функционирования волосковых клеток критический внутриулитковый потенциал, наступает глухота.
7. Синдром Брауна-Виалетто-Ван Лаере. Это редкое заболевание неизвестной этиологии, принадлежащее, как считается, к группе заболеваний двигательных нейронов с понтобульбарным параличом и глухотой. Двусторонняя сенсоневральная глухота может сопровождаться различными расстройствами черепных нервов; обычно страдает двигательный компонент нижних черепных нервов, но могут поражаться и спинномозговые двигательные нервы и верхние двигательные нейроны. Отмечаются как семейные, так и спорадические случаи. Результаты некоторых нейрофизиологических исследований указывают на поражение нервов с последующим улучшением, что подтверждает обусловленность заболевания первичным поражением нерва, а не патологическими изменениями двигательных нейронов (Degrandis, 2005; Prabhu, 2005).
8. Синдром Вольфрама. Синдром Вольфрама — аутосомно-рецессивное заболевание, характеризующееся несахарным диабетом, сахарным диабетом, атрофией зрительного нерва и глухотой, возникающих при мутации WFS1. Отмечается потеря восприятия высоких частот, может также развиваться периферическая нейропатия с атонией мочевыводящих путей и психическими расстройствами. Доминантная низкочастотная сенсоневральная потеря слуха также может вызываться мутациями WFS1 (Lesperance, 2003). Поскольку обычное скрининговое исследование слуха новорожденных не идентифицирует потерю восприятия частот ниже 2000 Гц, дети с риском развития глухоты при синдроме Вольфрама нуждаются в специальном наблюдении.
9. Врожденная фиксация основания стремечка с истечением перилимфы. Этот Х-сцепленный синдром проявляется смешанной или сенсоневральной глухотой, кондуктивный компонент которой обусловлен врожденной фиксацией основания стремечка. При попытках его мобилизации возникает профузное истечение эндолимфы. На КТ выявляются расширение внутреннего слухового прохода с наличием аномальных путей сообщения между субарахноидальным пространством и эндолимфой улитки. У женщин-носителей может наблюдаться легкая потеря слуха и нетяжелые аномалии внутреннего уха. Локус картирован на Xq21.1; было продемонстрировано, что во многих семьях мутация вызвана делецией, иногда могут быть вовлечены близлежащие гены, что ведет к умственной отсталости и хороидеремии.
10. Синдромальная потеря слуха, сопутствующая дерматологическим расстройствам. Существует по крайней мере пять дерматологических расстройств, которым сопутствует синдромальная потеря слуха; они также вызывают различные по тяжести поражения других органов. Примером таких расстройств является синдром Барта-Памфри, являющийся аутосомно-доминантным расстройством, характеризующимся сенсоневральной потерей слуха, пальмоплантарной кератодермой, утолщением межфаланговых суставов и лейконихией (Richard et al., 2004).
11. Доминантная атрофия зрительного нерва, сенсоневральная потеря слуха, птоз и офтальмоплегия. Этот синдром вызывается миссенс-мутацией в ОРА1 (Payne et al., 2004). ОРА1 является нуклеарным геном, но генный продукт локализуется в митохондриях; это свидетельствует о том, что митохондриальная дисфункI щя может быть частой причиной развития многих форм синдромальной и несиндромальной атрофии зрительного нерва, потери слуха и наружной офтальмоплегии.
12. Синдромы краниофациальных аномалий. Значительное число синдромальных состояний с краниофациальными аномалиями связано с глухотой, для некоторых из них описаны молекулярные нарушения, вызывающие патологию слуха. Примером такого состояния является бранхио-оторенальный синдром, связанный с мутациями генов EYA1 и EYA4, поражающих факторы транскрипции. При этом синдроме может развиваться гипоплазия улитки типа Мондини с гипоплазией и смещением слуховых косточек. При УЗИ выявляется агенезия, гипоплазия или дисплазия почек.
У детей с глубокой/прогрессирующей потерей слуха и кранифациальными аномалиями высока вероятность выявления отклонений при КТ. Расширение вестибулярного водопровода коррелирует с прогрессирующими нарушениями слуха. Аномалии каменистой части височной кости при КТ выявляются у 6,8-12,8% пациентов с двусторонней сенсоневральной потерей слуха и у 30% кандидатов на установку кохлеарных имплантов.
При дисплазии типа Мондини отмечается улитка с нормальным базальным завитком и дистальным мешком. При синдроме CHARGE (Colobomata — колобома, Heart defect — порок сердца, Atresia of the choanae — атрезия хоан, Retarded growth/development — задержка poста/развития, Genital hypoplasia—гипоплазия гениталий, Ear anomalies or deafness — аномалии уха или глухота) и синдроме Ватер-Рападилино (синдром Ватер — сочетание вертебральных дефектов, атрезии/стеноза ануса, трахео-эзофагеальной фистулы, дефектов лучевой кости и аномалий почек; в то время как синдром Рападилино включает дефекты лучевой кости, отсутствие/гипоплазию надколенника, расщелину неба, диарею и вывихи суставов, маленький рост, длинный тонкий нос, нормальный интеллект и потерю слуха) полукружные каналы отсутствуют или недоразвиты (Bamiou et al., 2000).
13. Сенсоневральная потеря слуха при хромосомных аномалиях. Хромосомные аномалии наблюдаются примерно у 5% детей с сенсоневральной глухотой. Hultcrantz (2003) сообщил, что 61% женщин с синдромом Тернера страдали средним отитом и сенсоневральным снижением слуха; иногда эти изменения отмечались еще с шестилетнего возраста и прогрессировали со временем.
6q минус синдрому может сопутствовать двусторонняя тяжелая сенсорная глухота. Это редкое расстройство, вызываемое трисомией или моносомией 6q, приводит к умственной отсталости, микроцефалии, асимметрии лица, широкой переносице, гипертелоризму, эпикантусу, косоглазию, высокому изогнутому небу, дефекту межжелудочковой перегородки и припадкам. Также описаны тетраплегия и грыжа диафрагмы (Schuster et al., 2003).
б) Потеря слуха ребенком при неврологических заболеваниях. Leuzzi et al. (2000) описал врожденную патологию белого вещества с атаксией, лейкодистрофией, и сенсоневральной потерей слуха, к возрасту 12 лет прогрессирующей в полную глухоту. Описана семейная мозжечковая атаксия с гипергонадотропным гипогонадизмом и сенсоневральной глухотой, но глухота обычно развивается во взрослом возрасте (Storey, 2001; Georgopoulos et al., 2004).
У детей с аутизмом наблюдается повышенная частота нарушений слуха, по данным одной группы глубокая двусторонняя потеря слуха наблюдалась в 3,5% случаев (Rosenhall et al., 1999). Часто встречается вызываемый звуками дискомфорт— 18% случаев; серозный средний отит наблюдается в 23,5%; с сопутствующей кондуктивной потерей слуха в 18,3%.
Частота сенсоневральной потери слуха при синдроме Ретта в исследовании Pillion et al. (2003) оценивалась в 17,3%, она чаще встречалась у детей старшего возраста и пациентов, получавших противосудорожную терапию.
Сенсоневральная потеря слуха может сопутствовать нейродегенеративным расстройствам, таким как синдром Гунтера, и сенсорно-моторной нейропатии при атаксии Фридриха и синдрому Шарко-Мари-Тута.
1. Болезнь Рефсума. Болезнь Рефсума характеризуется пигментным ретинитом, аносмией, хронической сенсорно-моторной нейропатией и атаксией. Потеря слуха встречается часто, поэтому пациенты, жалующиеся на нарушения слуха, должны проходить полное аудиометрическое обследование (Bamiou et al., 2003). При этом заболевании развивается разная степень потери слуха — от легкой, влияющей преимущественно на восприятие высоких частот, до умеренной; некоторые данные указывают на то, что может развиваться скрытое поражение слухового нерва. Oysu et al. (2001) описал отсутствие слуховых вызванных потенциалов ствола (СВПС) при наличии отоакустической эмиссии при болезни Рефсума с потерей слуха.
Это указывает на то, что потеря слуха может быть вторичной по отношению к нейропатии слухового нерва, с нарушениями на уровне выше наружных волосковых клеток. В этой связи возможно ограничение преимуществ и появление риска шумового поражения наружных волосковых клеток при использовании слуховых аппаратов, последнее нужно обязательно учитывать и отоакустическая эмиссия должна выполняться всем пациентам с болезнью Рефсума.
2. Митохондриопатии. Была описана косегрегация мутаций митохондриальной ДНК A1555G и G4309A, приводящая к глухоте и митохондриальной миопатии. Симптомы включают в себя прогрессирующую наружную офтальмоплегию, непереносимость физической нагрузки и глухоту, вызываемую приемом аминогликозидов (Campos et al., 2002). Митохондриальные цитопатии могут проявляться различными симптомами, но иногда сенсоневральная потеря слуха является первым проявлением; описан хороший эффект кохлеарных имплантов у некоторых пациентов.
Сенсоневральная глухота — частый симптом у пациентов с миоклонической эпилепсией с рваными красными мышечными волокнами (MERRF). В этой ситуации однотональная пороговая аудиометрия может выявить двустороннюю сенсоневральную потерю слуха; первичное поражение, как оказывается, локализуется в улитке, хотя в некоторой степени могут поражаться и ретрокохлеарные структуры (Tsutsumi et al., 2001).
Высокая частота (42%) сенсоневральной потери слуха у детей с митохондриальными энцефалитами, включая синдром Кирнса-Сейра, атаксию Фридриха и MELAS (митохондриальная энцефалопатия, лактацидоз и инсультоподобные эпизоды). Нарушение слуха прогрессирует, но не имеет прогностического значения для оценки течения основного заболевания. Получены данные, указывающие, что могут поражаться кохлеарные и ретрокохлеарные структуры (Zwirner и Wilichowski, 2001). Гистопатологические исследования при MELAS выявляют тяжелую дегенерацию сосудистой полоски и дегенеративные изменения клеток спирального ганглия, вызывающих сенсоневральную потерю слуха (Karkos et al., 2005).
3. Недостаточность биоптерина. У трех четвертей пораженных младенцев развивается потеря слуха, которая может быть тяжелой и сохраняться после начала терапии (Wolf et al., 2002). Потерю слуха можно полностью предотвратить при постановке диагноза до развития симптомов и назначении заместительной терапии биотином.