Мутации, поражающие факторы транскрипции, внутриклеточные белки, трансмембранные белки, внеклеточные белки и энергопродукцию способны привести к нарушению слуха (полный обзор см. Nance, 2003).
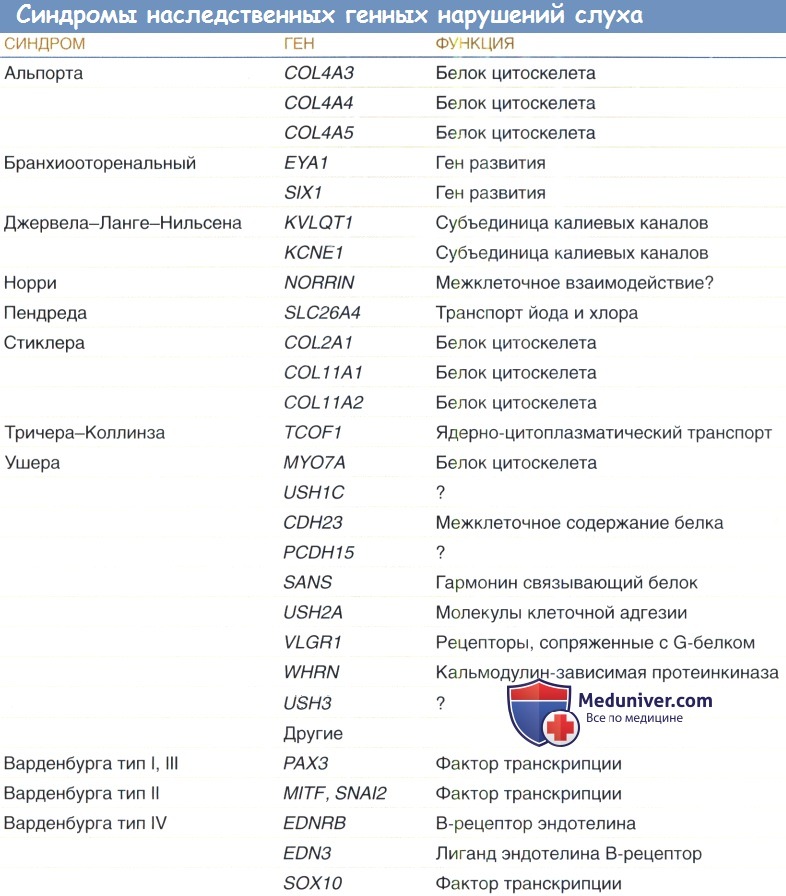
Мутации, поражающие факторы транскрипции, могут вызывать тяжелые формы глухоты. Дефекты генов РАХЗ, MITF и SOX10 являются причиной различных форм синдрома Ваарденбурга. Мутации, поражающие гены POU4F3 и POU3F4, приводят к доминантной форме прогрессирующей потери слуха и Х-сцепленному синдрому врожденной фиксации основания стремечка. Бранхио-ото-ренальный синдром является результатом мутаций, поражающих гены EYA1 и EYA4.
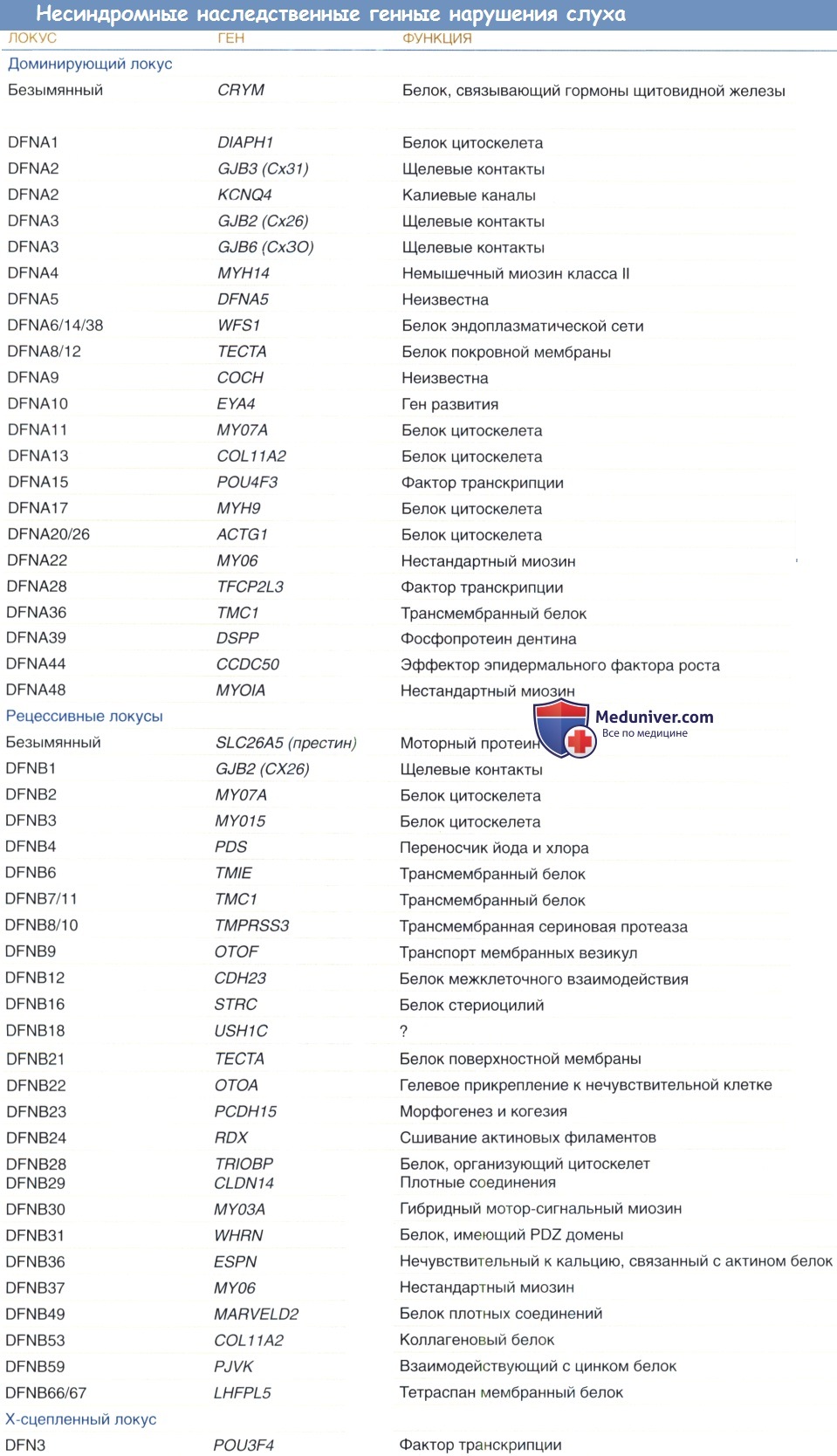
Мутации, поражающие внутриклеточные белки, могут вызывать образование атипичных миозинов; различные мутации MYO7A могут вызывать как доминантный, так и рецессивный синдром Ашера; мутации, поражающие MYO6 и MYH9, приводят к прогрессирующим формам доминантной потери слуха; и дефекты MYO15 вызывают врожденную форму глубокой рецессивной потери слуха.
Мутации, вызывающие образование аномальных структурных протеинов, как, например, вызываемые дефектами DIAPH1 и STRC, приводят к прогрессирующей потере слуха с доминантным механизмом наследования. Экспрессия этих генов особенно сильна в волосковых клетках, где они участвуют в полимеризации актина и продукции стереоцилина, являющихся компонентами протеинов микроворсинок.
Мутации, поражающие ген TCOF1 на 5q31, вызывают синдром Трешера Коллинза.
Мутации, поражающие трансмембранные протеины, могут привести к каналопатиям, и эти мутации вызывают нарушения минимум в трех белках семейства коннексинов, формирующих щелевой контакт: СХ26, СХ30 и СХ43. Два гена калиевых каналов, KBLQT1 и KCNE1, поддерживают нормальный гомеостаз кохлеарной эндолимфы, дефекты этих генов вызывают две формы рецессивного синдрома Ервелла-Ланге-Нильсена. Дефект SLC26A4, кодирующего связанный с мембраной протеин, участвующий в ионном транспорте, является причиной синдрома Пендреда.
CDF123 на 10q21 входит в семейство кальций-зависимого семейства генов кадгеринов, являющихся медиаторами клеточной адгезии; известно, что аномалии этого гена приводят к глухоте.
Мутации, поражающие три гена коллагена, COL2A1, COL1A2 и COL11А1, приводят к аномалиям внеклеточных протеинов и вызывают три описанные формы синдрома Стиклера; мутации COL4A5, COL4A3, COL4A4 приводят к развитию аутосомного и сцепленного с полом синдрома Альпорта.
Наконец, потерю слуха могут вызывать и нарушения энергопродукции. Так как нуклеарные и цитоплазматические гены активны в митохондриях в области оксидагивного фосфорилирования, потеря слуха может быть частью множества комплексных неврологических синдромов, вызванных делецией митохондриальной ДНК или точечными мутациями, поражающими митохондриальные тРНК. Также пептид слепоты/дистонии, являющийся продуктом гена Xq22, вызывает глухоту, слепоту, отсталость и дистонию при синдроме Транебьерга.