Нервная система при нарушении обмена гликопротеинов (маннозидозе, фукозидозе, галактосиалидозе)
Данные заболевания имеют общие характеристики с муколипидозами. Сиалидозы I и II типа относятся к обеим группам. Описано три основных заболевания.
а) Маннозидоз. Маннозидоз связан с дефицитом альфа-маннозидазы А и В, вызванным несколькими мутациями с геном альфа-маннозидазы (Beccari et al., 2003), приводящими к накоплению богатых маннозой олигосахаридов. Тяжелые формы заболевания (I тип) характеризуются началом в детском возрасте, Гурлер-подобным внешним видом, задержкой умственного развития, утратой слуха и гепатоспленомегалией. II тип характеризуется началом в подростковом возрасте и более мягким течением, но отмечаются значимые совпадения между проявлениями обеих форм (Bennet et al., 1995). Когнитивный дефицит имеет вариабельный характер и обычно не прогрессирует (Noll et al., 1989).
б) Фукозидоз. Причиной фукозидоза является дефицит фермента фукозидазы, кодируемого геном FUCA1; описано множество мутаций данного гена (Lin et al., 2007), приводящих к накоплению фукозы в тканях. Описано три типа заболевания: 1 тип или тяжелая форма с началом в раннем детском возрасте, 2 тип с началом в возрасте около 18 месяцев и редкий 3 тип с началом в подростковом возрасте. 2 тип встречается чаще всего и характеризуется медленным течением.
Клинически заболевание проявляется скелетными аномалиями, кожными изменениями (диффузной ангиокератомой туловища, затрагивающей преимущественно гениталии и десны) и неврологическими симптомами, часто в виде дистонии (Gordon et al., 1995). Часто встречаются промежуточные формы заболевания (Willems et al., 1991). В качестве лечения проводится трансплантация костного мозга.
в) Галактосиалидоз. Галактосиалидоз развивается в результате сочетания дефицита нейраминидазы и бета-галактозидазы из-за отсутствия антитела, предотвращающего протеолиз активного фермента. Заболевание имеет генетически гетерогенный характер (Suzuki et al., 1988).
Начало ювенильной формы заболевания приходится на возраст от 5 до 10 лет и проявляется мозжечковыми и экстрапирамидными знаками. Миоклонические припадки и миоклонус действия обычно формируется через несколько лет, а когнитивная деградация начинается поздно. Костные аномалии не развиваются, за исключением частого расширения первого поясничного позвонка. Постепенно формируются грубые черты лица. Детская форма заболевания может походить на ганглиозидоз I типа (Galjaard et al., 1984).
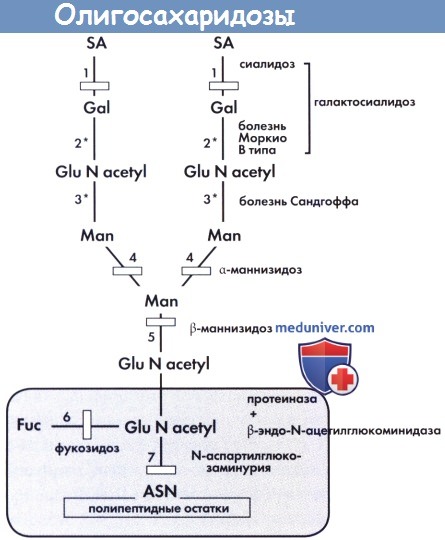
Опигосахаридозы: катаболизм гликопротеинов. Две части цепи распадаются после действия протеиназ и β-эндо-N-ацетилглюкоминидазы.
Сокращения: 1 — сиалидаза—а-нейраминидаза. 2 — β-галактозидаза.
1+2 — галактосиалидозы. 3 — гексозаминидаза А или В. 4 —- α-маннозидаза.
5 —β-маннозидаза. 6 — α-фукозидаза. 7 — N-аспартилглюкозаминидаза.