Наследственная тирозинемия I типа (HTI) характеризуется прогрессирующей печеночно-почечной недостаточностью и накоплением тирозина и его метаболитов. При острой и хронической формах большинство пациентов без лечения умирает от печеночной недостаточности или гепатомы. Тем не менее, у младенцев и детей причиной смерти независимо от печеночной недостаточности может быть острая периферическая нейропатия.
Данный вид нейропатии, описанный в основном среди пациентов из Квебека, может возникать независимо от этнического происхождения (Ashorn et al., 2006).
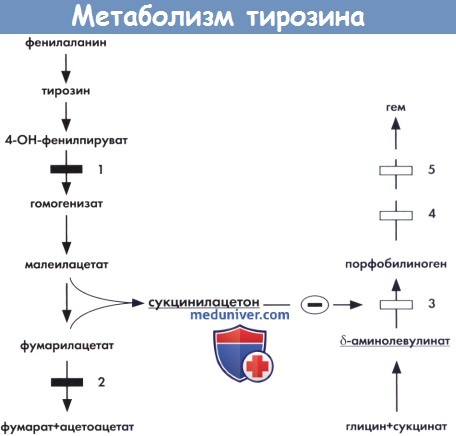
а) Биохимические и генетические изменения. Тирозинемия I типа (HTI) является аутосомно-рецессивным заболеванием, вызванным дефицитом фумарилацетоацетазы, последнего фермента, участвующего в катаболизме тирозина. Малеилацетоацетат и фумарилацетоацетат накапливаются и метаболизируются с образованием сукцинилацетона. Сукцинилацетат ингибирует дегидротазу дельта-аминолевулината (АЛДГ) и является причиной аномально высокой экскреции дельта-аминолевуленовой кислоты (δ-АЛК) с мочой. Накопление сукцинилацетона и δ-АЛК в плазме и моче позволяет диагностировать заболевание, для подтверждения которого измеряется активность фумарилацетоацетазы в лимфоцитах или фибробластах или же проводится молекулярный анализ.
Антенатальная диагностика основана на сходных анализах амниотической жидкости или образцов ворсин хориона. Тирозинемия I типа (HTI) в настоящее время расценивается как заболевание, отвечающее критериям скрининга новорожденных (Magera et al., 2006).
Метаболизм тирозина и его взаимосвязь с синтезом гемма.
1 — пара-ОН-фенилпируватдиоксигеназа (участок ингибирования нитизиноном).
2 — фумарилацетоацетаза. 3—6 - аминолевулинатдегидротаза
4 — острая интермиттирующая порфирия. 5 — другие порфирии.
б) Патогенез. Патогенез изучен не полностью. По неясным причинам не у всех пациентов развиваются неврологические кризы. Сравнение с другими патологическими состояниями с повышением δ-АЛК, такими как острая интермиттирующая порфирия, наследственный дефицит АЛДГ (Jaffe и Stith, 2007) и отравление свинцом, позволяет предположить, что рецидивирующее начало нейропатии связано с накоплением δ-АЛК и/или дефицитом печеночного гема (Lancet, 1990).
Трансплантация печени и лечение нитизиноном, позволяющие корректировать накопление δ-АЛК, предотвращают неврологические кризы (Mitchell et al., 1990; Gibbs et al., 1993).
в) Клинические проявления. Симптомы могут начинаться в течение нескольких недель (острые формы) или месяцев (подострые формы) после рождения или в последующие годы (хронические формы). У пациентов отмечаются признаки печеночной и почечноканальцевой недостаточности (синдром де Тони-Дебре-Фанкони). При острых и подострых формах печеночная недостаточность может быть основным проявлением, а почечноканальцевая недостаточность проявлялась только биологически. В отсутствие лечения смерть от почечной недостаточности наступает в течение нескольких месяцев. При хронических формах печеночная недостаточность выражена слабее, а поражение почек проявляется в виде почечноканальцевой недостаточности с гипофосфатемическим рахитом. Гепатома и терминальная стадия почечной недостаточности являются поздними осложнениями.
Неврологические кризы, описанные как симптомы, подобные порфирии, наряду с интеркурентными инфекциями могут возникать у пациентов с острыми и подострыми формами заболевания. Проявления включают стремительно прогрессирующую диффузную боль, локализующуюся преимущественно в области ног и живота, сопровождающуюся рвотой, раздражительностью, гипертонией, болезненными признаками опистотонуса и живыми сухожильными рефлексами. В дальнейшем может развиться стремительный восходящий паралич в сочетании с арефлексией, приводящий к дыхательной недостаточности и смерти при отсутствии искусственной вентиляции.
При развитии описанных симптомов пациент остается в сознании. Самоповреждения полости рта, такие как прикусывание языка и стирание и выпадение зубов, отмечаются преимущественно в начальной фазе заболевания.
Могут развиваться припадки, вторичные по отношению к метаболическим нарушениям (гипонатриемии, гипоксии) и стойкой гипертензии. Длительность выздоровления варьирует от случая к случаю. Чаще всего отмечается полное восстановление, но множественные рецидивы приводят к хронической инвалидизирующей периферической нейропатии с амиотрофией. ЭЭГ и состав спинномозговой жидкости не изменяются. При последовательном исследовании проводимости двигательных и чувствительных нервов было продемонстрировано прогрессирующее повреждение двигательных потенциалов действия и скорости проводимости нерва с гистопатологической дегенерацией аксонов и демиелинизацией (Mitchell et al., 1990; Gibbs et al., 1993).
Описано два случая, когда у молодых пациентов на момент диагностики на МРТ отмечалась аномальная миелинизация белого вещества головного мозга. У одного из них отмечалось два аномальных пика при протонной магнитно-резонансной спектроскопии данных повреждений, которые, вероятно, были связаны с молекулами тирозина. Годы спустя у второго пациента, страдающего хронической формой заболевания, были выявлены двусторонние изменения бледного шара без каких-либо других повреждений головного мозга (Sener, 2005).
г) Лечение тирозинемии I типа (HTI). Нитизинон является сильным ингибитором действия парагидроксифенилпируват диоксигеназы, второго этапа метаболизма тирозина. Введение данного препарата пациентам с тирозинемией предотвращает накопление токсических метаболитов с дальнейшим усилением тирозинемии. Усиление тирозинемии можно предупредить соблюдением диеты с ограничением фенилаланина/тирозина. Такой режим дает выраженный положительный эффект. Лечение нормализует функцию печени и почек и предотвращает неврологические кризы.
Тем не менее, для подтверждения безопасности и эффективности (в отношении предотвращения формирования гепатокарциномы) нитизинона при длительном применении требуется больше информации (Koelink et al., 2006; McKiernan, 2006).