Нервная система при фенилкетонурии и гиперфенилаланинемии
Гиперфенилаланинемия (ГФА) представляет собой повышение уровня фенилаланина в крови натощак по сравнению с показателями, выявляемыми у здоровых пациентов идентичного возраста. Гиперфенилаланинемия (ГФА) является группой заболеваний, среди которых наиболее частым является дефицит фенилаланингидроксилазы (ФАГ). Небольшое количество случаев связано с дефектом системы кофактора биоптерина (Burgard et al, 2000).
а) Биохимические и генетические изменения при фенилкетонурии и гиперфенилаланинемии:
1. Дисфункция метаболизма. Для гидроксилирования фенилаланина до образования тирозина необходимо три фермента: ФАГ, карбиноламиндегидротаза (КАД) и дигидропротеинредуктаза (ДПР), и два кофактора: тетрагидробиоптерин (ТГБ) и редуцированный НАД. На основании уровня фенилаланина в плазме и остаточной активности ФАГ в печени выделено три наследственных фенотипа ГФА в связи с дефицитом ФАГ: классическая фенилкетонурия, атипичная фенилкетонурия и нефенилкетонурическая ГФА. Дефекты синтеза и метаболизма 5,6,7,8-тетрагидробиоптерина (ТГБ) являются причиной ГФА и нарушений обмена нейротрансмиттеров.
2. Генетические изменения. ГФА является преобладающим нарушением метаболизма аминокислот среди представителей белой расы, частота заболевания составляет приблизительно 1 на 10000 живых новорожденных. Данное аутосомно-рецессивное заболевание вызвано более чем 500 мутациями локуса ФАГ. Различные группы мутаций преобладают в определенной этнической популяции, что позволяет проводить пренатальную диагностику, выявлять носителей и прогнозировать фенотип фенилкетонурии, связанный с определенным гаплотипом.
Некоторые мутации ФАГ приводят к дефициту ФАГ с остаточной ферментной активностью, которая усиливается под действием ТГБ. В таких случаях фармакологические дозы ТГБ приводят к минимум 30% снижению уровня фенилаланина в крови (Fiori et al., 2005).
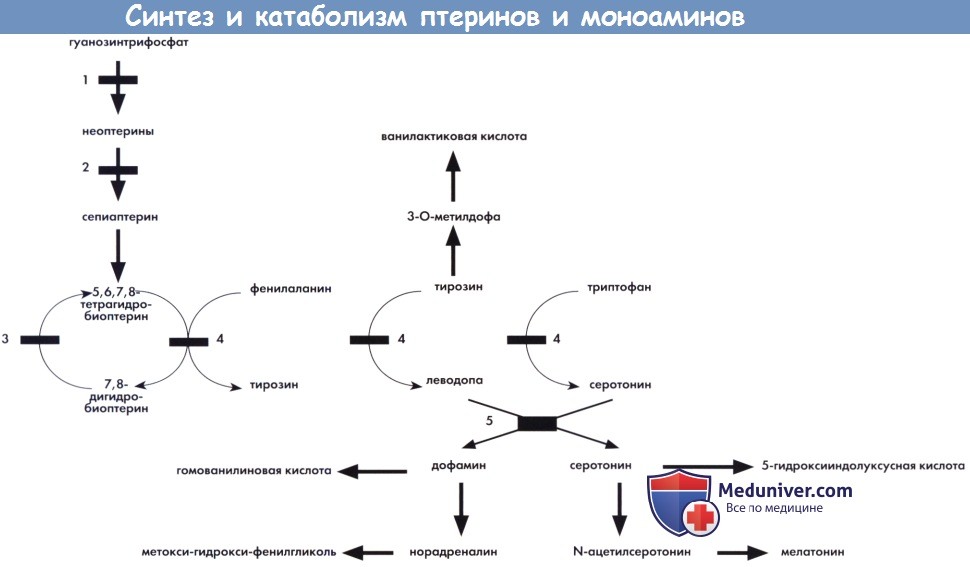
Синтез и катаболизм птеринов и моноаминов
1 — гуанозинтрифосфатциклогидролаза. 2 — биоптеринсинтаза. 3 — дигидроптеридинредуктаза.
4 —фенилаланин-, тирозин-, триптофангидроксилазы.
5 — ароматическая L-декарбоксилаза аминокислот (зависимая от пиридоксальфосфата).
3. Патогенез. Считается, что клинические проявления гиперфенилаланинемии (ГФА) являются результатом накопления фенилаланина и его вторичного воздействия на химические процессы в головном мозге. Фактически, фенилкетонурия чаще всего сопровождается задержкой умственного развития, в то время как при нефенилкетонурической гиперфенилаланинемии умственное развитие не меняется, что предполагает наличие порогового уровня фенилаланина во внеклеточных жидкостях, при превышении которого персистирующая постнатальная (или фетальная) гиперфенилаланинемия приводит к необратимому повреждению головного мозга. В случае если пороговая величина достигается позже, после прекращения соблюдения диеты пациентами, которым ранее проводилось лечение фенилкетонурии, развиваются обратимые химические изменения, способные оказывать воздействие на нейрофизиологические функции.
У пациентов с фенилкетонурией при высоком уровне фенилаланина в плазме отмечается снижение синтеза нейротрансмиттеров. Дефект синтеза нейротрансмиттеров может быть связан с конкурентным ингибированием транспорта крупных аминокислот (тирозина, триптофана и разветвленных цепей аминокислот) в головной мозг через гематоэнцефалический барьер и из спинномозговой жидкости обратно в кровь, что приводит к низкой концентрации тирозина и триптофана в головном мозге пациентов, несмотря на высокое содержание данных веществ в спинномозговой жидкости, а также, возможно, с конкурентным ингибированием гидроксилирования тирозина высоким уровнем фенилаланина.
В головном мозге пациентов старшего возраста, которым не проводилось лечения фенилкетонурии, отмечается аномальная миелинизация, уменьшение массы мозга и снижение содержания миелина. Данное пагубное влияние подтвердилось в ходе исследования на мышах (модель НРН-5). Результаты настоящих или ранее проведенных наблюдений стали основой для гипотезы о снижении количества миелина вследствие ингибирования специфичной для олигодендритных клеток АТФ-сульфурилазы, приводящего к низкому содержанию сульфатидов в миелине, который в свою очередь подвергается протеолитическому распаду. В дальнейшем отмечается потеря нейронов и уменьшение количества межнейронных связей, что было продемонстрировано путем количественной оценки плотности рецепторов нейротрансмиттеров. Если перенести на человека результаты, полученные в ходе исследований на животных, особое поражение гиппокампа и затылочного пространства могут объяснить некоторые нейрофизиологические нарушения у пациентов с фенилкетонурией, которым не проводилось лечения или лечение было недостаточным.
Аномальный синтез белков головного мозга вследствие дезагрегации полисом и снижении скорости удлинения полипептидной цепи могут приводить к снижению массы головного мозга. Дезагрегация полисом отмечается также в сердце и головном мозге плодов крыс, подвергающихся воздействию ГФА беременной самки; данное обстоятельство имеет отношение к фетопатии, связанной с ГФА беременной женщины.
Снижение содержания ДНК и синтеза в нейронах, подвергающихся воздействию фенилаланина в высоких концентрациях, также может объяснять снижение пролиферации нейронов, потерю нейронов и нарушение роста головного мозга.
б) Клинические проявления. Фенилкетонурия при отсутствии лечения Клинические проявления фенилкетонурии, по поводу которой не проводилось лечения, включают задержку умственного развития, неврологические аномалии и вненеврологические симптомы (хотя сроки их возникновения варьируют у различных пациентов). Задержка умственного развития часто сочетается с микроцефалией. Аномалии ЭЭГ встречаются часто (78-95% случаев), но только у 25% пациентов отмечаются припадки, чаще всего генерализованные. Нередко встречается психотическое поведение с гиперактивностью, деструктивным и самодеструктивным поведением, импульсивностью и неконтролируемым поведением с эпизодами эмоционального возбуждения.
У большинства пациентов слабопигментированная кожа с экзематозными проявлениями. Общее физическое развитие обычно хорошее.
Клинический фенотип представляет преимущественно исторический интерес, так как в настоящее время симптомы предотвращаются за счет ранней диагностики и лечения. Тем не менее, до сих пор отмечаются случаи пропущенной ГФА у новорожденных в случае, если тесты не проводились или были получены ложноотрицательные результаты.
в) Фенилкетонурия, по поводу которой проведено раннее лечение. У детей с фенилкетонурией, выявленной при рутинном скрининге новорожденных, по поводу которой вскоре после рождения была начата диетотерапия с ограничением фенилаланина, отмечается нормальный уровень умственного развития (Hanley, 2004). Тем не менее, результаты ретроспективных исследований свидетельствуют о том, что даже при максимально благоприятных условиях лечения у детей отмечается тенденция к более низкому коэффициенту IQ, чем у родственников первой линии, и худшей способности к обучению, чем у сибсов и детей контрольной группы. Часто встречаются субклинические нейрофизиологические (изменения вызванных потенциалов и проводимости нерва) и нейропсихологические нарушения, особенно у пациентов, не строго соблюдающих диету.
Частота аномалий на ЭЭГ увеличивается с возрастом вне зависимости от раннего и строгого соблюдения диеты, а результаты МРТ свидетельствуют о том, что дисмиелинизация является практически универсальным проявлением среди пациентов с классической и атипичными формами фенилкетонурии. Изменения на МРТ затрагивают затылочно-теменные области и в наиболее тяжелых случаях распространяются на лобные и височные доли. Описанные изменения не имеют явной взаимосвязи ни с клиническими или нейропсихологическими проявлениями, ни с контролем потребления фенилаланина в раннем детском возрасте, но коррелируют с уровнем фенилаланина в крови на момент проведения нейровизуализации и частично обратимы при снижении концентрации фенилаланина в крови.
г) Лечение фенилкетонурии и гиперфенилаланинемии (ГФА). У пациентов с классической и атипичной фенилкетонурией для предотвращения необратимых повреждений головного мозга диета с исключением фенилаланина должна быть начата вскоре после рождения. Ежедневное пероральное применение тетрадигидробиопентина может быть альтернативой диете у пациентов с чувствительной к тетрадигидробиопентину формой атипичной фенилкетонурии (Muntau et al., 2002). Для предотвращения интеллектуальной, неврологической и нейрофизиологической деградации после расширения диеты универсальной тактикой является пожизненное лечение. У пациентов с нефенилкетонурической ГФА (уровень фенилаланина в сыворотке <600 ммоль/л) неблагоприятных исходов не отмечается и необходимость соблюдения диеты отсутствует.
В случае начала лечения в позднем детском возрасте возможно некоторое улучшение поведения, но результаты длительного наблюдения предполагают большую подверженность изменениям поведения после прекращения соблюдения диеты, чем среди пациентов, которым проводилось раннее лечение.
д) Фенилкетонурия при беременности. Синдром фенилкетонурии матери является фетопатией, которая развивается у детей, рожденных женщинами с фенилкетонурией, по поводу которой не проводилось лечение, несмотря на то, что у самих детей обычно фенолкетонурия отсутствует (Lee et al., 2005).
1. Фенотип. Данный синдром характеризуется микроцефалией, низкой массой тела при рождении, дисморфизмом, врожденными дефектами и дальнейшей задержкой развития. Имеются признаки, что повышенный риск непосредственно коррелирует с уровнем фенилаланина в крови матери во время беременности, явный пороговый эффект отсутствует. Когда концентрация фенилаланина в плазме превышает 1200 мкмоль/л (классическая фенилкетонурия) отмечается высокая частота патологии плода: в 92% — врожденное слабоумие, в 73% — микроцефалия, в 40% — внутриутробная задержка развития, в 12% — врожденная мальформация сердца. У детей, родившихся у матерей с атипичной фенилкетонурией (уровень фенилаланина в крови 600-1200 мкмоль/л) также отмечается повышенная частота микроцефалии, задержки умственного развития и других врожденных аномалий, таких как пороки сердца (Lenke и Levy, 1980).
В случае, если ГФА матери не превышает 900 мкмоль/л, врожденные аномалии встречаются редко, но сохраняется высокий риск микроцефалии и задержки умственного развития. Микроцефалия является наиболее постоянным клиническим проявлением, таким образом, наличие данного признака всегда предполагает ГФА матери, даже если у женщины нормальный интеллект. На МРТ отмечается аномальное развитие мозолистого тела без изменений белого вещества, характерных для детей с фенилкетонурией (Levy et al., 1996). У детей женщин с ГФА легкой степени (160-600 мкмоль/л) отмечается нормальный уровень раннего коэффициента IQ. Тем не менее, размер головы, масса при рождении и уровень интеллекта могут находиться в обратной зависимости от концентрации фенилаланина у матери; результаты исследований предполагают отсутствие порогового уровня фенилаланина при воздействии на головной мозг плода.
2. Патогенез. Поражение плода и постнатальная фенилкетонурия, вероятно, характеризуются сходным патогенезом, когда фенилаланин является инициатором повреждения. У животных всех видов концентрация аминокислот у плода выше, чем у самки (приблизительное соотношение составляет 1,48). Таким образом, «безопасный» для матери уровень фенилаланина может быть причиной «небезопасного» уровня фенилаланина у плода.
Фенилаланин, вероятно, соперничает с другими нейтральными аминокислотами при транспорте через плаценту и в головном мозге, что может привести к замедлению роста плода и аномальному созреванию центральной нервной системы. Дефицит тирозина и триптофана может играть центральную роль в данном процессе, так как варианты врожденных аномалий при фенилкетонурии матери характерны для повреждения производных нервного гребня (сердца, аортальной дуги и лица). Представляется вероятным, что нарушение нормальной миграции гребня и развития сочетается с недостаточным синтезом нейротрансмиттеров плода из тирозина и триптофана (Kudo и Boyd, 1996).
3. Профилактика. Имеются достоверные доказательства, что в случае контроля уровня фенилаланина в крови матери до зачатия и в течение беременности, исход для плода благоприятный. Рекомендуемый уровень фенилаланина в крови матери составляет 250-360 мкмоль/л. Таким образом, для предотвращения заболевания необходимо соблюдение строгой диеты с низким содержанием фенилаланина и адекватным содержанием калорий и питательных микроэлементов.