Нервная система при нарушении обмена аминокислот с разветвленными цепями (АКРЦ)
Болезнь кленового сиропа (БКС), метилмалоновая ацидурия, пропионовая ацидурия, изовалериановая ацидурия и другие вторичные по отношению к метаболизму АКРЦ органические ацидурии в совокупности составляют наиболее часто встречающиеся врожденные дефекты метаболизма аминокислот. Заболевания имеют много общих симптомов и обычно соответствуют одному из трех клинических типов; тяжелой форме у новорожденных, интермиттирующей форме с поздним началом и хронической прогрессирующей форме. Рецидивирующая кома (основное проявление данных заболеваний) связана с непосредственным токсическим воздействием накопившихся метаболитов, в то время как хроническое накопление может препятствовать развитию центральной нервной системы или метаболизму головного мозга, приводя к задержке развития (Morton et al., 2002; Deodato et al., 2006; Vockley и Ensenauer, 2006).
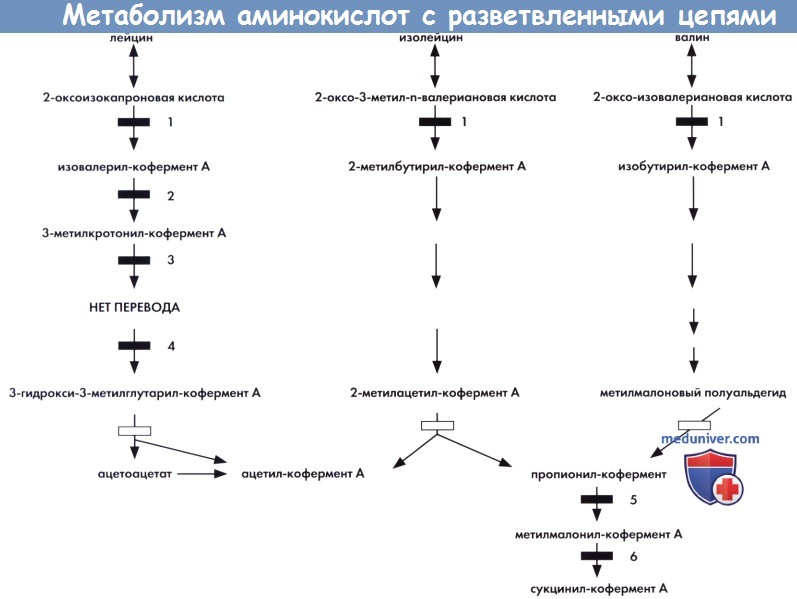
а) Биохимические и генетические изменения. Эти заболевания являются результатом дефекта ферментов, участвующих в метаболизме лейцина, валина и изолейцина. Три нейтральных АКРЦ изначально метаболизируются обычным путем: вслед за транасаминированием происходит тиамин-зависимое декарбоксилирование, дефицит которого отмечается при БКС (этап 1). Дефект декарбоксилирования является причиной накопления АКРЦ и соответствующих кетокислот. В дальнейшем пути катаболизма АКРЦ расходятся. Лейцин метаболизируется до ацетоацетата и ацетил-кофермента А. Специфическая ферментная недостаточность возможна на каждом этапе. Дефицит изовалерил-кофермент А дегидрогеназы является причиной изовалериановой ацидурии (этап 2), изовалерат является высокотоксичным веществом.
Дефицит 3-метилкротонил-кофермент А карбоксилазы, биотин-зависимого фермента, (этап 3) приводит к 3-метил-кротонил-глицинурии в результате мутации декарбоксилазы апофермента или аномального метаболизма биотина. 3-метилглутаконовая ацидурия I типа (этап 4) является другим редким заболеванием, вызванным дефектом катаболизма лейцина (Ly et al., 2003). Катаболизм валина и изолейцина протекает с образованием пропионил-кофермента А и матилмалонил-кофермента А. Метионин и треонин, жирные кислоты с нечетным количеством углеродных остатков и боковая цепью холестеринового радикала, являются другими предшественниками пропионил-кофермента А. Пропионил-кофермент А образует метилмалонил-кофермент А с участием биотин-зависимой декарбоксилазы (этап 5), дефицит которой вызван мутацией декарбоксилазы апофермента (пропионовая ацидурия) или аномальным метаболизмом биотина. Метилмалонил-кофермент А в два последовательных этапа под действием метил-кофермент А эпимеразы (Dobson et al., 2006), а затем под действием В12-зависимой метилмалонил-кофермент А мутазы (этап 6) превращается в сукцинил-кофермент А.
Дефицит активности каждого фермента приводит к метилмалоновой ацидурии. Аномальный метаболизм витамина В12 является причиной различных форм метилмалоновой ацидурии. До соединения пропионил-кофермента А описаны дефекты каждого этапа катаболизма изолейцина и валина, приводящие к ряду метаболических нарушений (Nguyen et al., 2002; Matern et al., 2003; Poll-The et al. 2004b; Salomons et al., 2007). Многие накапливаемые интрамитохондриальные метаболиты кофермента А являются субстратами эстерификации карнитина, процесса, который приводит к накоплению специфических ацилкарнитинов, выявляемых в цельной крови с помощью двойной масс-спектрометрии. Следовательно, большинство описанных заболеваний поддаются массовому неонатальному скринингу (Tarini, 2007).
Почти все заболевания данной группы наследуются аутосомно-рецессивным путем; дефицит 2-ме-тил-3-гидроксибутирил кофермент А дегидрогеназы является сцепленным с Х-хромосомой заболеванием. Антенатальная диагностика может проводиться путем оценки метаболитов в амниотической жидкости, определения активности специфических ферментов в образцах ворсин хориона и амниотических клетках и/или выявления патогенетических мутаций. По результатам программ скрининга новорожденных в сочетании с исследованиями мутаций было выявлено, что у ряда участвовавших в скрининге пациентов с изовалерьяновой ацидурией или 3-метилкротонглицинурией выявлялся слабо выраженный или даже бессимптомный фенотип.
б) Нейропатология. Имеющиеся результаты патологоанатомических исследований свидетельствуют об атрофии и различных гистологических изменениях мозга. Губчатая дегенерация белого вещества является неспецифическим повреждением, выявляемым у новорожденных. Часто описываемая у пациентов с метилмалоновой или пропионовой ацидурией дегенерация базальных ганглиев является заметным признаком среди детей старшего возраста с некрозом бледного шара, губчатыми и кистозными полостями. Серое вещество головного мозга и мозжечка и зубчатое ядро могут быть повреждены за счет диффузной вакуолизации вокруг нейронов и внутри нейтрофилов (Feliz et al., 2003). Субкортикальный отек головного мозга во время острой метаболической декомпенсации часто является причиной смерти у пациентов с БКС. Описано небольшое количество случаев кровоизлияний в мозжечок.
Патогенез поражения мозга при нарушениях обмена АКРЦ изучен плохо. В острой стадии заболевания накопление токсических метаболитов может быть причиной депривации энергии в связи с ингибированием определенных ферментов, участвующих в окислении пирувата в цикле Кребса или в митохондриальной дыхательной цепи. Восприимчивость базальных ганглиев к депривации энергии может объяснять их частичное поражение при таких заболеваниях, в особенности при метилмалоновой и пропионовой ацидуриях. При БКС высокий уровень лейцина может привести к дефекту синтеза катехоламина, вторичного по отношению к дефекту захвата головным мозгом нейтральных аминокислот и триптофана. Таким же образом взаимодействие метилмалоновой кислоты с сукцинатом может приводить к снижению синтеза гамма-аминомасляной кислоты (ГАМК). Необратимые изменения могут быть результатом взаимодействия с синтезом миелина вследствие дефекта транспорта всех нейтральных аминокислот (Schwab et al., 2006). Кроме того, накопленные пропионил-кофермент А и метилмалонил-кофермент А являются предшественниками синтеза жирных кислот с нечетным количеством углеродных остатков и длинноцепочечными жирными кислотами с метиловым радикалом. Их объединение в липидные слои может быть причиной аномального синтеза липидной мембраны в центральной нервной системе.
Метаболизм аминокислот с разветвленными цепями, свидетельствующий о происхождении некоторых известных наследственных метаболических нарушений.
1 — декарбоксилаза АКРЦ (болезнь кленового сиропа).
2 — изовалерил-кофермент А дегидрогеназа (изовалериановая ацидурия).
3 - 3-метилкротонил-кофермент А карбоксилаза β-метилкротонилглицинурия).
4 - 3-метилглутаконил-кофермент А гидратаза β-метилглутаконовая ацидурия I типа).
5 — пропионил-кофермент А карбоксилаза (пропионовая ацидурия).
6 — метилмалонил-кофермент А мутаза (метилмалоновая ацидурия).
в) Клинические проявления:
1. Тяжелые формы заболевания у новорожденных. Типичная клиническая картина представлена неуклонной деградацией без явных причин у новорожденных младенцев после бессимптомного периода, продолжающегося несколько дней. Первыми признаками является плохое сосание и трудности при кормлении с последующим развитием необъяснимой и прогрессирующей комы. В коматозном состоянии у большинства пациентов отмечается аксиальная гипотония с периферической дистонией, хореоатетозы, эпизоды опистотонуса и «боксирующих» и «велосипедных» движений конечностей. Судороги могут развиваться на более поздних стадиях заболевания. У больных детей может развиваться отек мозга с выбуханием родничка, что вызывает подозрения в наличии инфекции центральной нервной системы. На ЭЭГ часто отмечается паттерн вспышкаподавление. Биохимические аномалии включают метаболический ацидоз и кетонурию в сочетании с гипераммониемией и гиперлактацидемией. Общий краткосрочный прогноз улучшается. В дальнейшем, несмотря на лечение, часто возникают интеркуррентные эпизоды с клиническими проявлениями, сходными с проявлениями поздних интермиттирующих форм.
2. Интермиттирующие формы заболевания с поздним началом. Приблизительно у трети пациентов нарушения обмена АКДЦ отмечаются в детском и даже в подростковом или взрослом возрасте. Повторяющиеся приступы развиваются часто, хотя в промежутках пациент может казаться практически здоровым. Начало заболевания в большинстве случаев провоцируется состояниями, усиливающими белковый катаболизм (инфекцией, травмой и т.д.), или усиленным потреблением белка, но могут развиваться без явной причины. Рецидивирующие эпизоды комы или летаргии с атаксией являются основными проявлениями. Чаще всего кома не сопровождается другими аномальными неврологическими признаками. Кетоацидоз и различные аномалии уровня глюкозы в крови свидетельствуют о метаболическом характере заболевания. Тем не менее, у небольшого количества пациентов заболевание может проявляться гемиплегией и гемианопсией или признаками и симптомами отека мозга, имитирующими цереброваскулярные события или опухоли центральной нервной системы.
Часто отмечаются аномалии ЭЭГ: медленные дельтаволны, утрата нормального альфа-ритма, легкие неспецифические нарушения ритма или крупные аберрантные паттерны. Очаговая активность (при наличии) обычно локализуется в височной области.
Пациенты могут умирать во время эпизодов заболевания. У нескольких пациентов с БКС зарегистрированы случаи тяжелого отека головного мозга со сдавлением ствола мозга. По результатам диффузно-взвешенной визуализации и магнитно-резонансной спектроскопии данное осложнение является обратимым цитотоксическим отеком. Отек, усиливающийся при избыточной регидратации, тем не менее, может быть основной причиной смерти среди всех пациентов с нарушениями обмена АКРЦ, у которых развилась острая декомпенсация. Некоторые выздоравливают без последствий. У многих детей, выживших после тяжелых длительных или рецидивирующих метаболических нарушений, сохраняются повреждения головного мозга. Частым последствием обострений являются припадки. В младенческом и раннем детском возрасте припадки склонны к генерализации, преобладает миоклонический тип. В старшем детском возрасте часто встречаются тонико-клонические припадки или атипичные абсансы. При нейровизуализации обычно выявляется атрофия мозга и некоторая отсрочка миелинизации. При отсутствии лечения или недостаточном лечении БКС может сохраняться отек мозга. Чаще всего поражаются ствол мозга, базальные ганглии и затылочное перивентрикулярное и мозжечковое белое вещество.
У возрастающего количества пациентов с органическими ацидуриями и, в особенности, метилмалоновой и пропионовой ацидуриями отмечается острый прогрессирующий экстрапирамидный синдром вследствие двустороннего некроза базальных ганглиев. Чаще всего данные повреждения встречаются в бледном шаре, но могут поражать другие базальные ганглии (Chemelli et al., 2000).
3. Хронические прогрессирующие формы. Гипотония, мышечная слабость, неспецифическая задержка развития и припадки в редких случаях являются единственными проявлениями изовалериановой, метилмалоновой или пропионовой ацидурии. Чаще всего данные изменения сочетаются с симптомами со стороны пищеварения и питания и плохой прибавкой веса. С другой стороны, данные хронические неврологические формы могут быть проявлением 3-метилкротонглицинурии, которая характеризуется неподдающимися лечению припадками, гипотонией, микроцефалией и задержкой развития (при рано начинающихся формах) или метаболической лейкодистрофией с эпизодами, подобными болезни Рея (у детей старшего возраста). Кроме того, многие бессимптомные случаи были выявлены с помощью скрининга методом тандемной масс-спектрометрии среди новорожденных. 3-метилглутаконовая ацидурия I типа может проявляться легкими неврологическими нарушениями (Nguyen et al., 2002). Малоновая ацидурия может проявляться изолированной задержкой развития или задержкой развития в сочетании с припадками, мышечной слабостью и кардиомиопатией (Salomons et al., 2007). Описаны редкие случаи 2-метилбутирилглицинурии и изобутирилглицинурии, проявлявшиеся мышечной слабостью и некоторой задержкой умственного развития.
Тем не менее, подавляющее большинство недавно зарегистрированных пациентов выявлено путем систематического скрининга и не имеют симптомов заболевания (Li et al., 2003), несмотря на доказанные мутации соответствующих генов (Matern et al., 2003; Pedersen et al., 2006). Дефицит 2-метил-3-гидроксибутирил-кофермент А дегидрогеназы может быть причиной сцепленного с Х-хромосомой прогрессирующего нейродегенеративного заболевания второго года жизни с признаками, напоминающими последствия гипоксическо-ишемического повреждения головного мозга новорожденных (Poll-The et al., 2004b). Нарушения движений, такие как хорея, в сочетании с расстройствами интеллекта могут быть признаком прогрессирующих форм любой органической ацидурии.
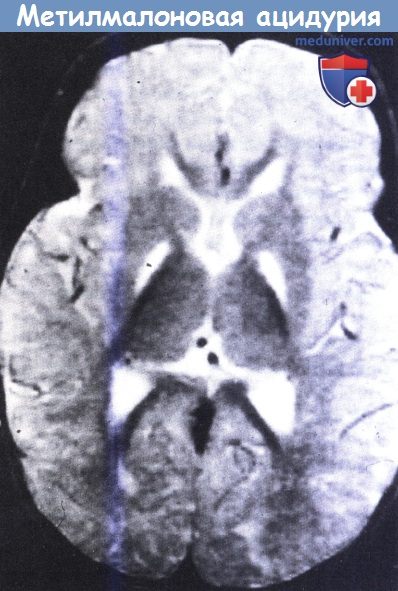
Метилмалоновая ацидурия.
На МРТ в Т2-режиме видны симметричные участки усиления сигнала в области бледного шара у восьмимесячного ребенка.
Также отмечаются признаки отсроченной миелинизации.
г) Биохимическая диагностика. Диагностика основана на выявлении аномальных промежуточных субстратов и сопутствующих веществ, образующихся в результате метаболического блока. У пациентов с БКС хроматография аминокислот характеризуется высоким уровнем лейцина, валина и изолейцина в плазме и моче, а также наличием аллоизолейцина. С другой стороны, все остальные блоки ферментов приводят к накоплению в плазме и экскреции с мочой органических кислот и диагностируются с помощью газожидкостной хроматографии и масс-спектрометрии, в то время как аминокислотная хроматография не позволяет поставить диагноз. Тандемная масс-спектрометрия позволяет диагностировать данные нарушения за счет выявления аномальных параметров ацилкарнитина, формирующихся в результате накопления специфических ацилкарнитинов. Для окончательной диагностики требуется оценка специфической ферментной активности в фибробластах или лейкоцитах и/или идентификация патогенной мутации в пораженных генах.
д) Лечение. Целью лечения является снижение количества токсических метаболитов (Ogier de Baulny et al., 2005). В периоде новорожденности в случае наиболее остро протекающего заболевания детям требуется активное питание, часто в сочетании с экзогенной детоксикацией. Долгосрочная диетотерапия предполагает ограничение потребления белка при сбалансированной по остальным компонентам диете. В связи с тем, что некоторые витамины являются кофакторами специфических ферментных превращений, очень большие дозы данных витаминов должны систематически тестироваться в каждом случае. Данное терапевтическое тестирование позволяет выявить пациентов, реагирующих на применение витаминов, у которых заболевание выражено в меньшей степени и необходима менее строгая диета. При лечении органических ацидурий систематически применяется поддерживающая терапия L-карнитином. При изовалериановой ацидурии и 3-метилкротонилглицинурии применение L-глицина и L-карнитина увеличивает экскрецию метаболитов с мочой и является эффективным средством детоксикации.
Несмотря на раннюю диагностику и лечение, в любом возрасте у таких детей сохраняется риск развития угрожающего жизни метаболического дисбаланса и неврологических последствий. Кроме того, у многих пациентов с метилмалоновой и пропионовой ацидурией отмечается прогрессирующая неврологическая дисфункция с нарушением интеллекта, атаксией, мышечной слабостью и недостаточностью питания. Другие осложнения затрагивают почки, поджелудочную железу и сердце. Учитывая данные долгосрочные осложнения, проводится трансплантация печени и почек. Трансплантация не способна предотвратить острую неврологическую деградацию и некроз базальных ганглиев (Leonard et al., 2001; Chakrapani et al., 2002).