Нервная система при врожденных нарушениях гликозилирования
Гликозилирование белков является необходимым этапом котрансляционного созревания растущей полипептидной цепи. Данный процесс необходим для жизнеспособности и нормального развития. Это касается большинства внеклеточных белков, таких как белки сыворотки (трансферрин, α1-антитрипсин, α1-антихимотрипсин, некоторые факторы свертывания, апо-липопротеин С-III), большая часть мембранных белков, таких как рецепторы специализированных клеток, и некоторые внутриклеточные белки, такие как лизосомальные ферменты. Врожденные нарушения гликозилирования были подразделены на нарушения N-и О-гликозилирования.
Нарушения, связанные с дефектами маннозо-а-О-гликозилирования сочетаются с синдромом Уокера-Варбурга и различными формами мышечных дистрофий, таких как болезнь мышц-глазмозга, врожденная дистрофия Фукуяма и сочетанные миопатии (описаны в других разделах) (Muntoni et al., 2004; Martin, 2005). Нарушения N-гликозилирования приводят к мультисистемным аномалиям, которые обычно включают поражение центральной нервной системы. Заболевания данной группы подразделяются на два типа. Тип I врожденных нарушений гликозилирования характеризуется дефектом элонгации гликана, в то время как тип II врожденных нарушений гликозилирования связан с дефектом путей процессинга (см. обзор в работах Leroy, 2006; Sparks, 2006).
а) Биохимические изменения и патогенез. Гликозилирование белков является очень сложным процессом, в котором участвует множество ферментов и транспортеров.
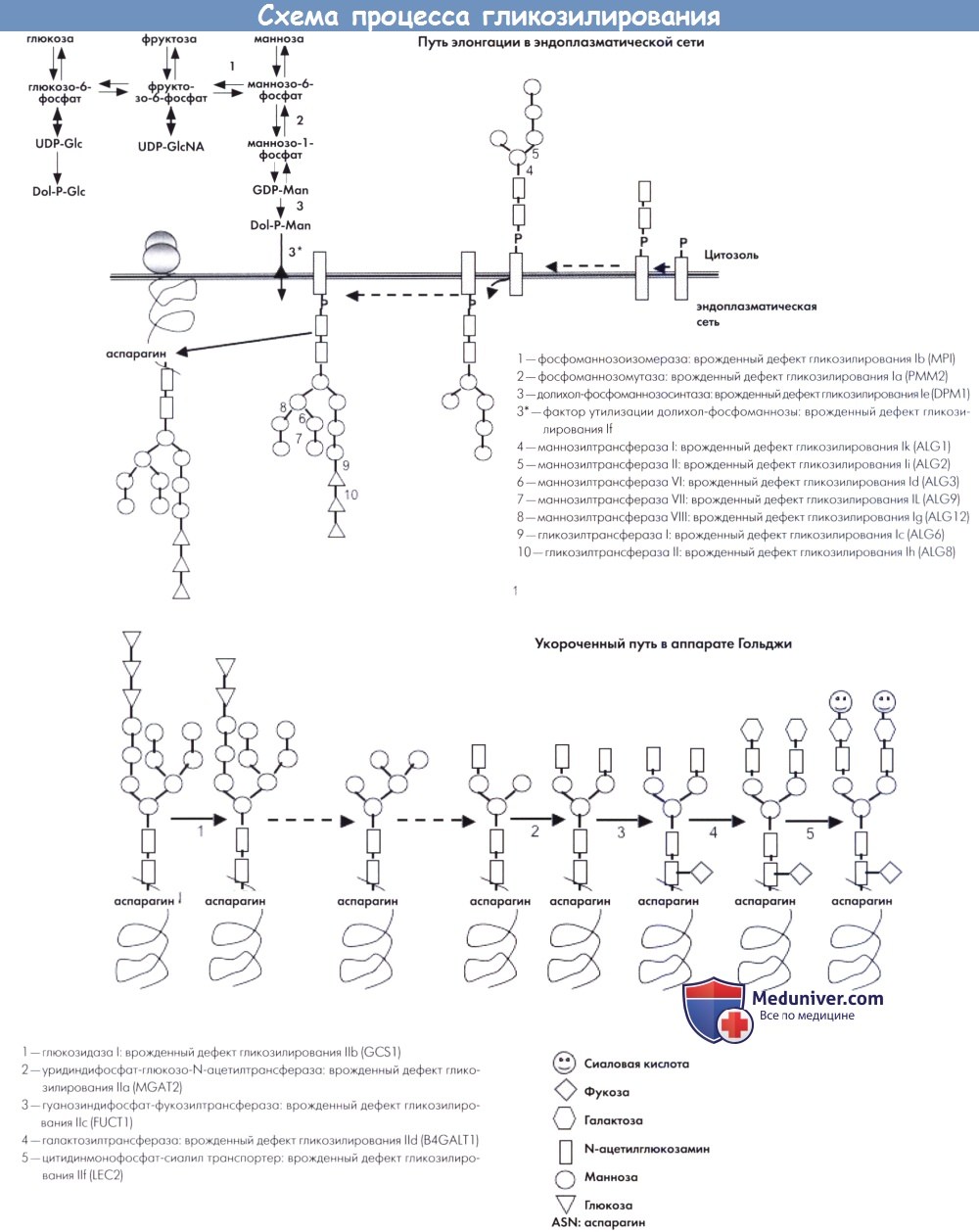
Формирование необходимых олигосахаридов, также называемых гликанами, происходит в эндоплазматической сети. Различные доноры сахаров активируются в качестве связанных с нуклеотидами сахаров и долихил-фосфатами моносахаридов на цитоплазматической стороне эндоплазматической сети. Синтез олигосахаридов инициируется прикреплением двух N-ацетил-глюкозамин-фосфатных единиц к липидному носителю (долихол-фосфату), необходимому для процесса элонгации олигосахаридов.
В дальнейшем ступенчатая элонгация происходит за счет прикрепления маннозных и глюкозных остатков. Каждый шаг катализируется специфическими трансферазами, которые кодируются соответствующими ALG генами. При последнем шаге олигозид переносится на образующийся белок с помощью олигосахарид-протеин-гликозилтрансферазы. Различные врожденные дефекты данного пути являются причиной врожденных нарушений гликозилирования I типа.
После переноса, опосредованного олигосахарид-протеин-гликозилтрансферазой, процесс продолжается в аппарате Гольджи в виде удавления глюкозных и маннозных остатков под действием специфических глюкозидаз и маннозидаз. На данном этапе процесс разделяется на два пути. Один путь заключается в лизосомальном фосфорилировании, а его дефекты являются причиной муколипидоза II типа (болезнь I клеток) и III типа (псевдо-Гурлер синдром). Второй путь обеспечивает специфическую ориентацию белков относительно участка их функционирования, например, внутри плазматической мембраны или для секреции в межклеточное пространство. Данный путь, также называемый укороченным, заключается в удалении маннозных остатков с помощью специфичной маннозидазы и прикреплении двух N-ацетил-глюкозамин-фосфатных единиц с помощью специфичных трансфераз.
В дальнейшем происходит прикрепление одной фукозы, двух сиаловых кислот и двух галактоз. Для данного пути созревания необходимы специфические транспортеры фукозы и сиаловых кислот и галактозилтрансфераза. Дефекты укороченного пути являются причиной врожденных нарушений гликозилирования II типа. Описано два других врожденных нарушения гликозилирования II типа, вызванные дефектами сохраненных олигомеров Гольджи. Сохраненный олигомер Гольджи представляет собой белковый комплекс, фиксированный на мембране Гольджи и участвующий в сохранении и функционировании аппарата Гольджи.
Схематическое описание процесса гликозилирования с указанием дефектов при врожденных нарушениях гликозилирования I и II типов:
(а) путь элонгации в эндоплазматической сети, (b) укороченный путь в аппарате Гольджи.
б) Диагностика. Диагностика врожденных нарушений гликозилирования проводится путем изоэлектрического фокусирования сывороточного трансферрина. Характеристики делятся на два типа. Тип 1 характеризуется повышением дисиало- и асиалотрансферриновых соединений одновременно со снижением тетрасиалотрансферрина. Тип 2 характеризуется увеличением трисиало- и моносиалотрансферриновых соединений. В дополнение к изофокусированию трансферрина изофокусирование других сиалиновых белков улучшает качество диагностики (Fang et al., 2004). Изоэлектрическое фокусирование аполипопротеина С-III используется для скрининга дефектов О-гликозилирования в отдельности или в сочетании с дефектами N-гликозилирования.
Выделяется три типа изоформ: аполипопротеин С-III0, аполипопротеин С-III1, аполипопротеин С-III2, в зависимости от количества остатков сиаловой кислоты, прикрепленных к гликопротеину (Wopereis et al., 2007).
в) Врожденные дефекты гликозилирования 1 типа. Врожденный дефект гликозилирования 1а типа (ВДГ-Iа) ВДГ-Ia является наиболее частым и наиболее изученным заболеванием из группы врожденных дефектов гликозилирования с частотой около 1 на 50000. Классические формы заболевания являются прогрессирующими и представлены тремя стадиями, развивающимися с возрастом (Hagberg et al., 1993; Stibler et al., 1994).
В младенческом периоде преобладают заметные мультисистемные поражения, которые прогрессируют от периода новорожденности до позднего младенческого возраста. Заболевание характеризуется плохим питанием, плохой прибавкой в весе и мышечной вялостью. Ранняя задержка психомоторного развития отмечается у всех пациентов в виде выраженной задержки развития крупной моторики, явной аксиальной гипотонии, мышечной слабости; позднее появляются признаки нарушения равновесия или истинной мозжечковой атаксии. Нормальная окружность головы при рождении может трансформироваться в приобретенную микроцефалию. Сухожильные рефлексы изначально слабые, арефлексия формируется в течение первых 2-3 лет жизни. Часто встречается зрительная невнимательность, блуждание взора и внутреннее косоглазие.
Врожденная катаракта встречалась в редких случаях. В большинстве случаев отмечался некоторый дисморфизм лица, втянутые соски и характерное строение тела с ограничением движений в суставах конечностей, деформацией грудной клетки и необычной липодистрофией со скоплением жира на ягодицах, в области промежности и/или на пальцах рук. Часто отмечаются липоатрофические полоски и бляшки. Слабо выраженная гепатомегалия сочетается с фиброзом печени или гигантоклеточным гепатитом. Отмечены случаи увеличения почек и протеинурии. Во многих случаях могут развиваться опасные эпизоды полиорганной недостаточности, которые включают тяжелые инфекции, печеночную недостаточность, выпот в перикард с тампонадой сердца и ступор, в отдельных случаях сочетающийся с припадками и внутричерепным кровоизлиянием. Данные острые эпизоды ухудшения являются причиной высокой детской смертности (15-20%).
В детском возрасте отмечается непрогрессирующая задержка умственного развития различной степени. У большинства пациентов коэффициенты DQ/IQ составляют около 50. Мозжечковая атаксия и периферическая нейропатия, способствующая мышечной атрофии, в большинстве случаев препятствуют самостоятельному передвижению. Возможны рецидивирующие эпизоды дискинетических или хореоатетозных движений. Обычно развивается косоглазие и пигментный ретинит. Инсультоподобные эпизоды, связанные с частичным тромбозом мозговых сосудов, развиваются у половины пациентов в возрасте после 4-5 лет и проявляются комой, припадками и транзиторной слепотой. Приблизительно у половины пациентов формируется эпилепсия.
В подростковом и взрослом возрасте состояние стабилизируется. Большинство пациентов овладевает рядом социальных навыков, но остаются иждивенцами.
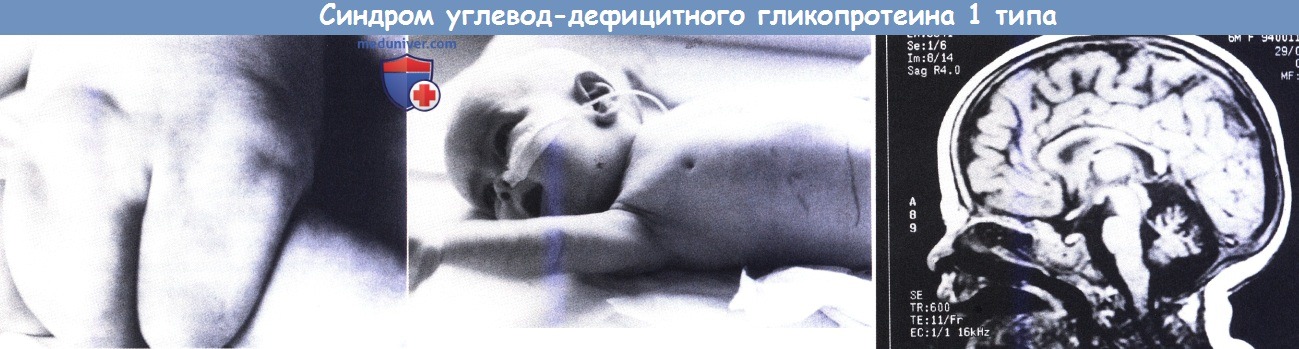
Синдром углевод-дефицитного гликопротеина 1 типа.
Слева: Аномальное отложение жира на ягодицах.
В центре: Втянутые соски; также отмечается затруднение питания (питание через зонд).
Справа: МРТ в сагиттальной проекции—заметная атрофия мозжечка и относительно тонкий ствол мозга.
Описанное течение заболевания касается большинства пациентов, тем не менее, описаны фенотипы с легкими или необычными проявлениями (Neumann et al., 2003; Coman et al., 2005; Noelle et al., 2005).
На ранних стадиях возможно умеренное повышение содержания белка в спинномозговой жидкости. Параметры электроэнцефалограммы неспецифичны. При электроретинографии отмечается прогрессирующее снижение ответа сетчатки (Andreasson et al., 1991). Зрительные, слуховые стволовые и соматосенсорные вызванные потенциалы могут отличаться от нормы. Скорость проводимости нерва снижена в моторных и сенсорных нервах, постепенное снижение отмечается вплоть до подросткового возраста.
При нейровизуализации у большинства пациентов в течение первого года жизни выявляется стремительно прогрессирующая гипоплазия мозжечка и ствола. Супратенториальные структуры обычно не отличаются от нормы, тем не менее, у трети пациентов отмечается центральная и/или корковая атрофия. Практически всегда отмечается оливопонтоцеребеллярная атрофия и миелиноподобные накопления в лизосомах (Eyskens et al., 1994). У двух близнецов, умерших в возрасте 4 месяцев и 6 лет, атрофия была более выражена при большей продолжительности жизни, что предполагает прогрессирование дегенеративного процесса. Очаговые инфаркты головного мозга предполагают окклюзию сосудов. При микроскопии выявляется полная утрата клеток Пуркинье, почти полная утрата гранулярных клеток в коре мозжечка и глиозядра моста (Stromme et al., 1991).
При биопсии икроножного нерва были выявлены аномальные слои миелина (Nordborg et al., 1991); отмечены лизосомальные накопления в клетках передних рогов (Eyskens et al., 1994), а также гепатоцитах, что свидетельствует об изменениях метаболизма миелина.
г) Другие врожденные нарушения гликозилирования 1 типа. Выявлено еще 11 вариантов нарушения путей элонгации олигосахаридов. Данным заболеваниям названия присвоены в алфавитном порядке (Ia—II) в соответствии с порядком описания. Аномалии развития и функции центральной нервной системы отмечаются при всех вариантах нарушений, за исключением одного (врожденное нарушение гликозилирования Ib). Врожденное нарушение гликозилирования-Ic является вторым по частоте заболеванием с клиническим фенотипом, сходным с врожденным нарушением гликози-лирования-Ia. Другие клинические фенотипы в настоящее время недостаточно описаны в связи с небольшим количеством пациентов в каждой группе. Некоторые характеризуются задержкой развития (от легкой до умеренной) с гипотонией, атаксией, косоглазием, нистагмом и припадками, варьирующими от фебрильных эпизодов до эпилепсии. У большинства пациентов отмечается тяжелая энцефалопатия новорожденных в сочетании с признаками, напоминающими врожденное нарушение гликозилирования-Iа.
У некоторых пациентов отмечается нейросенсорная тугоухость, слепота, колобома радужки и катаракта. Среди экстраневрологических симптомов часто встречается плохая прибавка веса, поражение скелета, кардиомиопатия или перикардиальный выпот, фиброз или цирроз печени и почечная недостаточность. В некоторых случаях выявляется водянка плода. Результаты МРТ могут быть нормальными или свидетельствовать о корковой и подкорковой атрофии, гипоплазии мозолистого тела или гипомиелинизации. Описаны случаи гипоплазии мозжечка, но данное проявление не является обязательным.
Врожденные нарушения гликозилирования 1 типа могут приводить к повреждению различных сывороточных гликопротеинов, оценка которых может способствовать диагностике. Низкое содержание свертывающих факторов и ингибиторов (фактора XI, антитромбина III, протеина С, протеина S, кофактора гепарина) может служить объяснением инсультоподобных эпизодов. В связи с неспецифическим просачиванием в сыворотку отмечается повышение содержание нескольких лизосомальных (например, аурилсульфатазы А, бета-гексаминидазы) и нелизосомальных ферментов (например, трансаминаз). Изменения также затрагивают многие транспортные белки (например, апопротеин В), гликопротеиновые гормоны (например, инсулиноподобный фактор роста-1) и факторы комплемента. Окончательный диагноз врожденного дефекта гликозилирования-1 подтверждается наличием сывороточных изоформ трансферрина 1 типа.
Учитывая данное обстоятельство, наиболее распространенные врожденные дефекты гликозилирования-Ia, Ib и Iс обычно диагностируются с помощью ферментных и молекулярных методов исследования. Другие типы заболевания диагностировались по результатам исследований, когда другие методы оказались неэффективны.
Врожденные нарушения гликозилирования I типа являются аутосомно-рецессивными заболеваниями, связанными с нарушением этапов сборки олигосахаридов. Все известные дефектные ферменты или транспортеры кодируются соответствующими генами; патогенетические мутации идентифицированы. Данная информация позволяет осуществлять достоверную пренатальную диагностику путем анализа мутаций в хорионической ДНК.
Эффективного лечения не существует.
д) Врожденные дефекты гликозилирования 2 типа. Врожденные дефекты гликозилирования 2 типа являются группой заболеваний, при которых затронут укороченный путь гликозилирования в аппарате Гольджи.
Врожденный дефект гликозилирования 2а типа Врожденный дефект гликозилирования 2а типа был первым описанным заболеванием данной группы. У небольшого количества пациентов отмечалась тяжелая задержка развития, отсутствие периферической нейропатии и нормальное строение мозжечка на МРТ. У пациентов отмечалась гипотония с младенческого возраста, генерализованные припадки с детского возраста и аномальное поведение со стереотипными движениями в виде потирания рук, давления языком и ударов головой. Все пациенты характеризовались низким ростом, скелетными деформациями, гипогонадизмом и небольшой печеночной недостаточностью. С биохимической точки зрения отмечается углеводный дефицит трансферрина со изоформой трансферрина 2 типа. Склонность к кровотечениям объясняется снижением содержания фактора свертывания XI. Недостаточность активности уридиндифосфат-глюкозо-N-ацетилтрансферазы II связана с мутациями гена MGAT2, расположенного на 14q21 хромосоме (Cormier-Daire et al., 2000).
Другие врожденные дефекты гликозилирования 2 типа Четыре других типа были связаны с дефектом действия глюкозидазы (IIb), галактозилтрансферазы (IId) и фукозы или транспортеров сиаловых кислот (IIc, IIf). Каждая группа заболеваний была представлена небольшим количеством или даже одним пациентом. Неврологические симптомы имели выраженный характер при IIb и IId типах заболевания и проявлялись в виде тяжелой энцефалопатии, некупируемых припадков и нормальной МРТ головного мозга (IIb) или умеренной задержки развития, мышечной слабости и мальформации Денди-Уокера (по результатам нейровизуализации) (IId). Тяжелые гематологические симптомы выявлялись при IIс и IIf типах заболевания. Результаты изоэлектрического фокусирования не отличались от нормы при всех типах заболевания, за исключением одного; врожденный дефект гликозилирования IIf типа сочетался с изменениями апопротеина-С-III1 (Wopereis et al., 2007).
Три других дефекта связаны с аномалиями комплекса сохраненного олигомера Гольджи (COG): CDG-IIe/COG7, CDG-IIg/COG1 и CDG-IIh/COG8. Результаты изоэлектрического фокусирования трансферрина плазмы 2 типа и свойств аполипопротеина-С-III0 свидетельствовали о повреждении N- и О-гликозилирования. Первое заболевание было описано у двух новорожденных сибсов, страдавших смертельной полиорганной недостаточностью и некупируемыми припадками. Дефект COG1 был выявлен у одного ребенка с дисморфическими проявлениями, некоторой задержкой развития и умеренной атрофией мозга и мозжечка при визуализации, выполненной в возрасте двух лет. У пациента с дефицитом COG8 отмечалась тяжелая энцефалопатия (Kranz et al., 2007).
Пероральное применение фукозы могло приводить к уменьшению выраженности гематологических изменений среди пациентов, страдающих врожденным дефектом гликозилирования Нс типа, но не оказывало воздействия на неврологический статус (Leroy, 2006). Для других типов заболевания специфического лечения не существует.
Ясно, что данная группа заболеваний недостаточно исследована и фенотипическая вариабельность среди уже описанных типов, а также дополнительные формы еще будут выделены после того, как станут известны все ферменты и соответствующие гены, задействованные в сложном процессинге гликозилирования.