а) Определения:
• Амелия: отсутствие одной конечности или более
• Микромелия: укорочение проксимального и дистального сегментов конечности
• Фокомелия: укорочение конечности, при котором кисть или стопа расположены вблизи туловища:
о Наиболее поражен, как правило, проксимальный сегмент
• Редукция конечности: отсутствие части скелета или мягких тканей конечности:
о Может быть продольной, поперечной или перемежающейся
• Гемимелия: отсутствие дистальной части конечности
б) Лучевая диагностика. Рекомендации по лучевой диагностике:
• Предпочтительный метод исследования:
о УЗИ в I и II триместрах:
- Возможна диагностика в I триместре с помощью ТВУЗИ
- В III триместре диагностика может быть затруднена
о 3D/4D УЗИ позволяет более детально изучить строение конечностей:
- Метод полезен при консультировании
• Советы по проведению исследования:
о Решающим признаком в дифференциальной диагностике является характер поражения конечностей:
- Симметричность/асимметричность поражения
- Поражение в большей степени верхних или нижних конечностей
- Наличие/патология кистей и стоп
- Пораженные сегменты конечностей
о Исключают сопутствующие аномалии, в частности орофациальные расщелины, торакоабдоминальные аномалии и пороки сердца:
- Синдромальная диагностика
о Обнаруживают проявления синдрома амниотических перетяжек
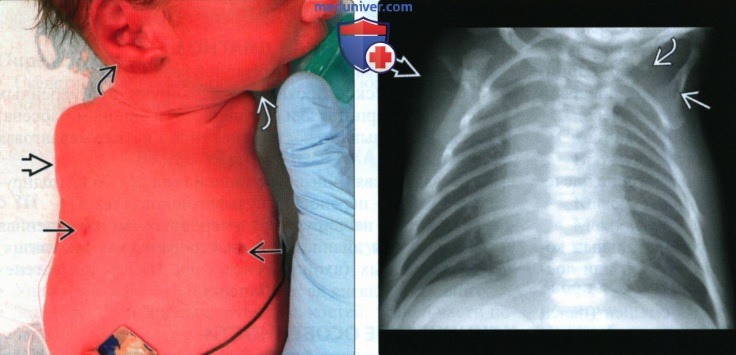
(Слева) Клиническая фотография. Доношенный новорожденный с амелией верхних конечностей в сочетании с фокомелией нижних конечностей. Микрогнатия. Ушные раковины увеличены, расположены низко и ротированы кзади. Гипоплазия и асимметричность сосков.
(Справа) Рентгенограмма того же новорожденного, прямая проекция. Верхние конечности отсутствуют. Лопатки и ключицы гипоплазированы.
в) Дифференциальная диагностика амелии, фокомелии у плода:
1. Амелия:
• Синдром Робертса/БС-синдром:
о Синдром Робертса и SC-фокомелию рассматривают как единое заболевание
о Клинические проявления:
- В 90% случаев - тетрафокомелия (в 10% случаев поражаются только верхние конечности)
- В 80% случаев - расщелина нёба
- Выраженная задержка роста плода и новорожденного
- Лицевая дизморфия
- Возможны аномалии мочеполовой системы, сердца, органов слуха и обоняния, синдактилия, переднее энцефалоцеле, микроцефалия
о Наследуется по аутосомно-рецессивному типу
о Большинство детей погибают внутриутробно или вскоре после рождения:
- Описаны редкие случаи более длительной продолжительности жизни
о Характерная цитогенетическая особенность - преждевременное разделение центромер:
- Образование пуфов на хромосомах с вторичными перетяжками в области центромер (1, 9, 16, короткое плечо акроцентрических хромосом, короткое плечо Y-хромосомы)
- Высокоспецифический признак синдрома Робертса
- Чувствительность в пренатальной диагностике неизвестна
- Сообщается о несовпадении результатов пре- и постнатальных исследований
• Тетраамелия:
о Встречается редко - 0,4:100 000 живых новорожденных
о Описаны сопутствующие аномалии:
- Агенезия и гипоплазия легких
- Орофациальные расщелины
- Отсутствие носа и ушных раковин
- Пороки сердца
- Аномалии мочеполовой системы, в том числе наружные половые органы промежуточного типа
- Атрезия ануса
- Эктодермальная дисплазия
о Часто обнаруживают сопутствующие аномалии внутренних органов
о Высокая перинатальная летальность
о Аутосомно-рецессивная тетраамелия связана с мутациями гена WNT3
• Изолированная фокомелия/амелия:
о Поражена одна конечность или более
• Амниотические перетяжки:
о Асимметричные: поражена одна конечность или более
о Орофациальные расщелины причудливой формы и аномалии черепа:
- Линии расщелин не соответствуют эмбриологическим линиям сращения
о Дефекты стенок туловища
о При УЗИ в околоплодных водах или при патологоанатомическом исследовании плаценты обнаруживают амниотические тяжи
о Круговые перетяжки конечностей и пальцев:
- Псевдосиндактилия
• Тромбоцитопения с отсутствием лучевой кости:
о Фокомелия верхней конечности, нередко выраженная
о В 50% случаев - аномалии нижних конечностей
о Гипомегакариоцитная тромбоцитопения
о Капиллярная гемангиома лица
о Аутосомно-рецессивный тип наследования
о Дифференциальная диагностика с анемией Фанкони:
- При TAR-синдроме типа 1 пальцы кисти не изменены
- В отличие от анемии Фанкони, TAR-синдром не связан с хромосомной нестабильностью
• DK-фокомелия:
о Синдром фон Фосс-Черствого
о Фокомелия, энцефалоцеле, тромбоцитопения, аномалии мочеполовой системы
о Аутосомно-рецессивный тип наследования
• Талидомидная эмбриопатия:
о Популярный в 1950-60-х годах в Европе седативный препарат; применялся для лечения утренней тошноты у беременных
о Отозван с рынка в 1962 г.: у детей, чьи матери на ранних сроках беременности получали талидомид, обнаруживали тяжелые пороки развития конечностей
о Механизм действия: нарушение ангиогенеза, воспалительный ответ
о Характерная совокупность аномалий: тетрафокомелия, пороки сердца, мочеполовой системы, лица и нервной системы
о В 1998 г. одобрен FDA в качестве препарата для лечения осложнений лепры:
- Экспериментальное лечение ВИЧ, язвенной болезни и воспалительных заболеваний
- Даже однократный прием препарата максимизирует риск эмбриопатии
2. Микромелия:
• Ахондрогенез:
о Задержка оссификации позвоночника
о Непропорционально крупная голова; оссификация в норме или снижена
о Короткие ребра ± переломы
о Микрогнатия
о Часто развивается водянка плода
о Тип 1А, 1В наследуются по аутосомно-рецессивному типу; тип 2 возникает случайно
• Ателостеогенез:
о Макроцефалия
о Микрогнатия
о Расщелина нёба
о Короткое туловище с гипоплазированной грудной клеткой, выпуклый живот
о Пальцы «автостопщика»
о Косолапость
о Укорочение трубчатых костей с расширением метафизов
о Широкая щель между I и II пальцами стопы
о Аутосомно-рецессивный тип наследования: диастрофическая дисплазия - мутации гена, кодирующего белок -переносчик сульфат-ионов (DTDST)
• Диссегментарная дисплазия:
о Тела позвонков неправильной формы с множеством ядер окостенения (анизоспондилия)
о Укорочение позвоночника, грудная клетка малых размеров с короткими ребрами
о Укорочение и утолщение седалищных и лобковых костей
о Укорочение, расширение и искривление трубчатых костей
о Аутосомно-рецессивный тип наследования
• Фиброхондрогенез:
о Расширение родничков и расхождение черепных швов, проптоз глазных яблок
о Расширение трубчатых костей, булавовидные эпифизы
о Задержка оссификации задних частей тел позвонков, передние расщелины позвонков
о Подвздошные кости расширены, медиальные и латеральные остеофиты
о Аутосомно-рецессивный тип наследования
• Несовершенный остеогенез II типа:
о Дефекты коллагена I типа (COL1A1, COL1A2)
о Выраженная задержка оссификации, в том числе черепа
о Множественные переломы ребер и трубчатых костей
о Возникает случайно; повторное возникновение связано с мозаицизмом гонад
• Синдром коротких ребер - полидактилии типа 1 и 3:
о Постаксиальная полидактилия
о Узкая грудная клетка и выпуклый живот
о Множественные аномалии внутренних органов, в том числе сердца, мочеполовой системы и лица
о Короткие трубчатые кости с зазубренными эпифизами
о Аутосомно-рецессивный тип наследования
• Группа диастрофической дисплазии:
о Включает диастрофическую дисплазию, ахондрогенез типа 1В и ателостеогенез типа 1
о Мутации гена DTDST
о Диастрофическая дисплазия:
- Прогрессирующий кифосколиоз
- Расщелина нёба
- Косолапость
- Пальцы «автостопщика»
- Высокий риск перинатальной смерти; при отсутствии тяжелых поражений позвоночника продолжительность жизни не снижается
- Аутосомно-рецессивный тип наследования
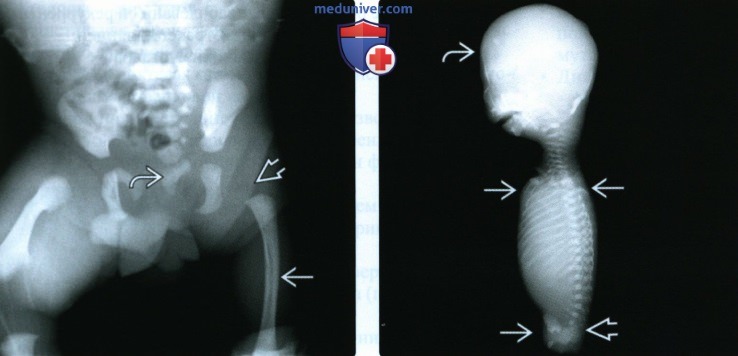
(Слева) Рентгенограмма новорожденного пациентки с неконтролируемым СД, прямая проекция. Сложный редукционный порок конечности: одностороннее отсутствие бедренной кости единственная трубчатая кость дистального сегмента. Дистальный отдел позвоночника и левая половина таза гипоплазированы.
(Справа) Рентгенограмма мертворожденного с тетраамелией, боковая проекция. Отсутствуют все конечности Непропорционально крупная голова, выраженная гипоплазия таза.
• Этиология:
о Аутосомно-рецессивная тетраамелия - мутации гена WNT3
о Диастрофическая дисплазия, ахондрогенез типа 1В, ателостеогенез типа 1 - мутации гена DTDST
о Несовершенный остеогенез II типа - мутации COL1A1, COL1A2
о Синдром Робертса (патология когезина) - мутации ESCO2
о Отдельные случаи изолированной фокомелии связаны с патологией ангиогенеза
• Генетические факторы:
о Синдром Робертса, TAR-синдром, DK-фокомелия: аутосомно-рецессивный тип наследования
о Микромелия: как правило, аутосомно-рецессивный тип наследования
о Повторные случаи тетраамелии в близкородственных браках: предположительно аутосомно-рецессивный тип наследования
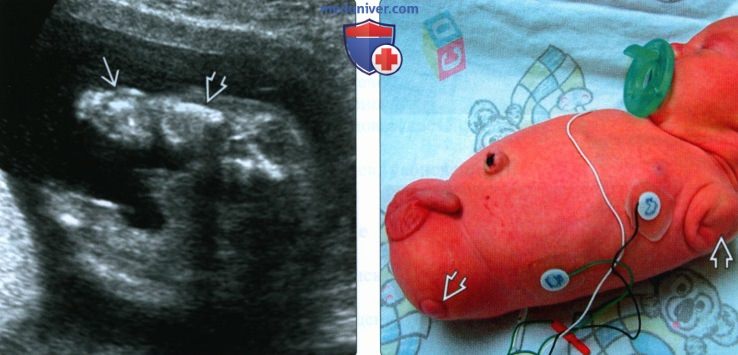
(Слева) УЗИ плода во II триместре. Ахондрогенез. Выраженная микромелия нижних конечностей. Отечность кожи связана с водянкой плода. Последняя находка характерна для многих летальных форм хондродистрофий.
(Справа) Клиническая фотография новорожденного с тетраамелией. На месте отсутствующих конечностей - кожные валики.
д) Клинические особенности:
1. Клиническая картина:
• Самые частые субъективные и объективные симптомы:
о Отсутствие или выраженное укорочение конечностей при УЗИ в I или II триместре
• Другие субъективные и объективные симптомы:
о Синдром Робертса: орофациальная расщелина и фокомелия/амелия
о Признаки скелетной дисплазии и микромелии
2. Естественное течение и прогноз:
• Большинство хондродистрофий с выраженной микромелией летальны в перинатальном периоде
3. Лечение:
• Пренатальное лечение отсутствует
• Выжившим требуется хирургическая коррекция прогрессирующих поражений позвоночника и конечностей
е) Особенности диагностики:
1. Важно знать:
• При подозрении на синдром Робертса показано цитогенетическое исследование - находят разделение хроматид в центромерном районе
2. Признаки, учитываемые при интерпретации результатов:
• Для дифференциальной диагностики важно установить вовлеченный сегмент и симметричность поражения конечностей
• Для исключения скелетной дисплазии изучают структуру костей, образующих конечность
• Исключают сопутствующие аномалии
ж) Список использованной литературы:
1. Haro Е et al: Sp6 and Sp8 transcription factors control AER formation and dorsal-ventral patterning in limb development. PLoS Genet. 10(8):el004468, 2014
2. Al-Qattan MM: WNT pathways and upper limb anomalies. J Hand Surg Eur Vol. 36(l):9-22, 2011
3. Eyaid W et al: A novel homozygous missense mutation (c.610G>A, p.Gly204Ser) in the WNT7A gene causes tetraamelia in two Saudi families. Am J Med Genet A. 155A(3):599-604, 2011
4. Goh ES et al: The Roberts syndrome/SC phocomelia spectrum -a case report of an adult with review of the literature. Am J Med Genet A. 152A(2):472-8, 2010