Данные по молекулярной биологии генетических заболеваний и синдромов, накопленные к настоящему времени, можно продемонстрировать на примере ключично-черепного дизостоза (OMIM: 119600) (рис. 104-106).
Ключично-черепной дизостоз - генерализованная дисплазия костей и зубов, наследуемая по аутосомно-доминантному типу (Mundlos, 1999; Baumert et al., 2006). Синдром имеет чрезвычайно вариабельный фенотип и различную пенетрантность патологического гена. При семейных формах заболевание может проявляться клинически с различной тяжестью (Golan et aL, 2003а; Baumert et al., 2005). Одним из характерных симптомов заболевания является гипо- или аплазия ключиц, которая делает возможным сведение плеч перед туловищем.
Несмотря на другие скелетные отклонения (Cooper et al., 2001), диагностируют ключично-черепной дизостоз прежде всего и главным образом по изменениям в зубочелюстной системе. Заболевание проявляется также задержкой окостенения черепа, очень большими размерами родничков и задержкой закрытия черепных швов. Наиболее очевидными симптомами являются наличие гиперкомплектных, часто ретенированных постоянных зубов, персистирование молочных зубов, гипоплазия средней части лица. Из других типичных черепно-лицевых изменений следует отметить также выдающийся лоб, частичное отсутствие костной перегородки носа и множественные вставочные (вормиевы) кости в затылочной области. Наблюдается также эбурнеация костей лицевого скелета, которые из-за глубокого склероза и утолщения напоминают слоновую кость, что затрудняет хирургическую коррекцию аномалии и способствует избыточному образованию рубцовой ткани после операции (Golan et al., 2003b).
В литературе описано свыше 1000 случаев, из которых более чем у 100 пациентов ключично-черепной дизостоз сочетался с другими аномалиями развития (Mundlos, 1999; Golan et al., 2003a). Заболеваемость составляет примерно 0,5 случая на 100 000 родившихся живыми детей в год (Lachman, 1996), а распространенность - 1 случай на 1 000 000 населения (Golan et al., 2003а). Аномалия примерно одинаково часто встречается среди представителей всех этнических групп, гендерная предрасположенность отсутствует.
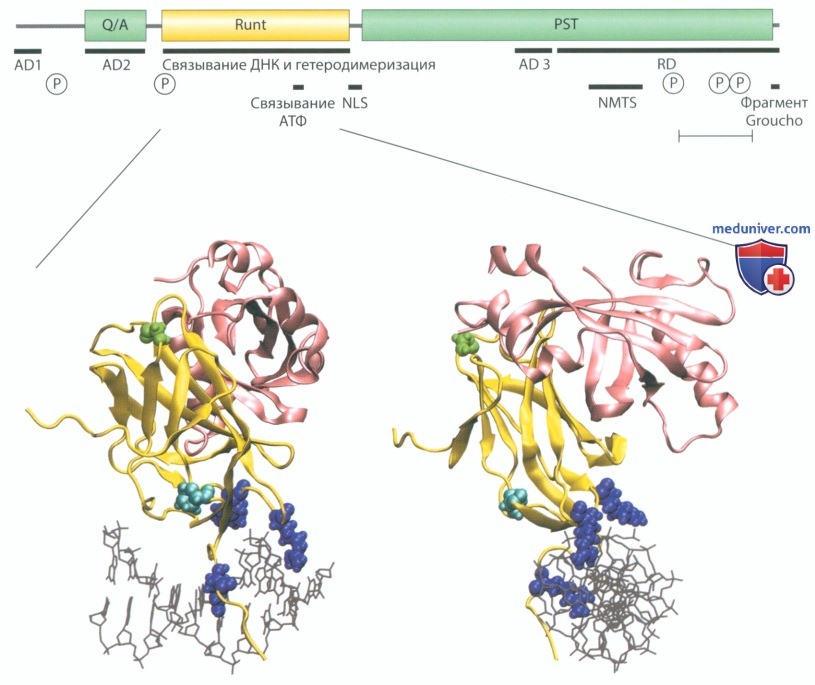
Рисунок 104. Генетическое заболевание: ключично-черепной дизостоз. На рисунке схематически представлены строение транскрипционного фактора RUNX2 (верхняя часть рисунка) и трехмерное изображение комплекса, образованного runt-доменом транскрипционного фактора RUNX2, корсвязывающим фактором β (CBFβ) и ДНК (нижняя часть рисунка).
RUNX2 имеет три крупных домена: Q/A, PST и runt. Q/A-домен (последовательность, содержащая множественные повторы глутамина и/или аланина) ответственен за транспорт белка в клеточное ядро, PST - за активацию и инактивацию транскрипции, runt - за связывание с ДНК. Транскрипционный фактор RUNX2 влияет на экспрессию других генов через активирующие домены AD1-AD3 или ингибирующий домен RD при участии дополнительных сигнальных молекул. Фосфорилирование («Р») и/или связывание с АТФ модулирует качество связи транскрипционного фактора RUNX2. После его синтеза в цитоплазме сигнальные последовательности NLS и NMTS направляют RUNX2 в клеточное ядро.
На трехмерных изображениях runt-домен (желтый цвет), ДНК (серый цвет) и CBFβ (розовый цвет) показаны сверху и повернуты на 90°. Шарики, окрашенные в разные цвета, представляют собой аминокислотные замены в мутировавших участках, выявленные у различных пациентов с ключично-черепным дизостозом. Синим цветом обозначен аргинин в ДНК-связывающем домене, бирюзовым - пролин, зеленым - мутационные аминокислотные замены в области гетеродимеризации с CBFβ.
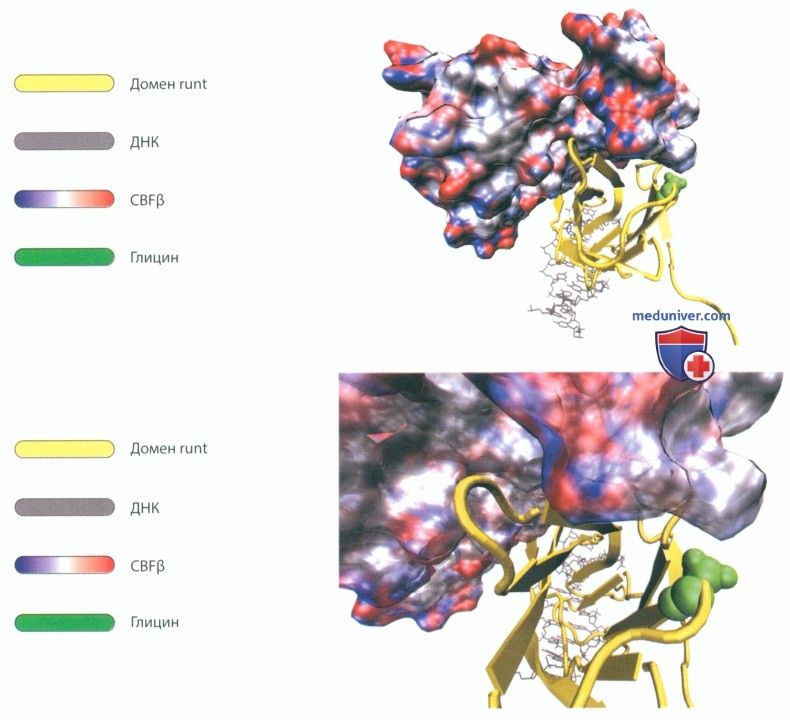
Рисунок 105. Генетическое заболевание: ключично-черепной дизостоз. Транскрипционный фактор RUNX2 играет важную роль в механотрансдукции, остеогенезе и морфогенезе кости. Он необходим для дифференцировки фибробластов.
На рисунке схематически показано трехмерное изображение комплекса, образованного ДНК, доменом runt и CBFβ. Последний представлен в виде поверхности, меняющей свой цвет в зависимости от заряда от красного (отрицательный заряд) через белый (электрически нейтральные участки) до синего (положительный заряд), что обусловлено природой аминокислотных остатков, образующих поверхность.
В немутировавшем транскрипционном факторе RUNX2 аминокислота глицин имеет строго определенную локализацию. Глицин среди аминокислот имеет наименьшие размеры, и к тому же у него нет заряда. Он располагается в непосредственной близости от места связывания β-субъединицы CBFβ.
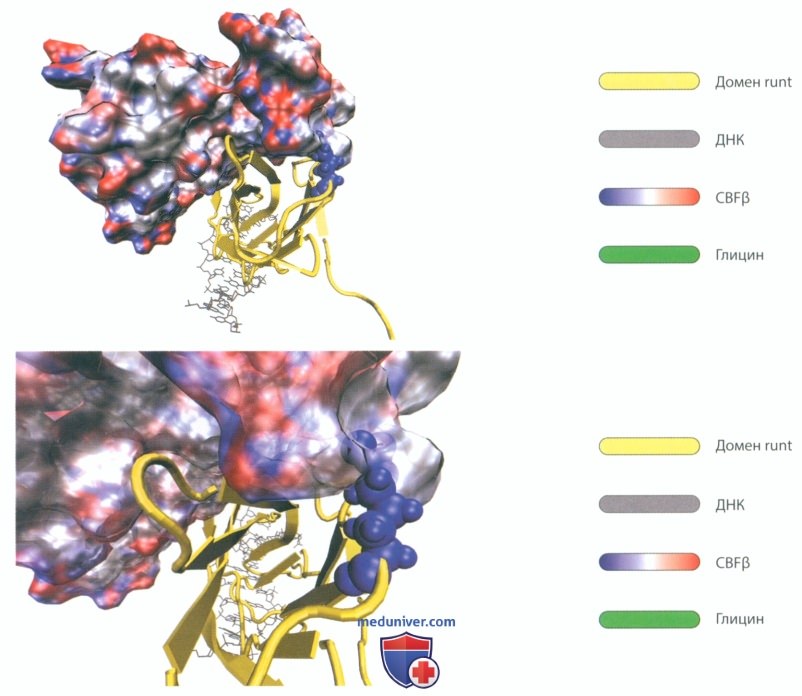
Рисунок 106. Генетическое заболевание: ключично-черепной дизостоз. Влияние мутации G146R на структуру и функцию транскрипционного фактора RUNX2.
Схематическое изображение комплекса, включающего в себя ДНК, домен runt и CBFβ. В результате мутации с несинонимичной заменой аминокислоты (миссенс-мутации) глицин замещен аргинином. Замена глицина на аргинин приводит к тому, что эта крупная аминокислота, несущая электрический заряд, выдается в область контакта и препятствует гетеродимеризации с CBFβ.
Несинонимичные точечные мутации сразу вызывают снижение или выпадение функции этого белка.
Генетической причиной ключично-черепного дизостоза являются гетерозиготные мутации гена RUNX2 (OMIM: 600211), который локализован на коротком плече хромосомы 6 (6р21) (Mundlos et al., 1997). Этот ген кодирует транскрипционный фактор RUNX2, который является одним из представителей семейства белков runt, наряду с факторами RUNX1 и RUNX3, играющими важную роль в развитии различных злокачественных опухолей (Ito, 2004). Транскрипционные факторы - это молекулы, оказывающие регулирующее действие на процесс считывания информации с ДНК (транскрипцию), иными словами, на трансформацию генетической информации в белок. Общим для runt-семейства транскрипционных факторов является чрезвычайно высокая вероятность наличия в их молекуле домена runt (Kagoshima et al., 1993) (рис. 104).
С помощью этого домена транскрипционный фактор RUNX2 связывается со специфическими, называемыми премоторными, участками гена и вместе с другими факторами регулирует транскрипцию. Исследования Komori и соавт. (1997) и Otto и соавт. (1997) показали, что транскрипционный фактор RUNX2 необходим для созревания остеобластов и что он играет ключевую роль в остеогенезе (Ducy et al., 1997). Mundlos и соавт. (1997) впервые описали мутации этого гена у больных с ключично-черепным дизостозом. Упомянутые выше функции транскрипционного фактора RUNX2 нашли отражение в структуре его молекулы, в которой имеются различные домены, ответственные за определенные функции, в частности за связывание с ДНК, за гетеродимеризацию или контроль специфического взаимодействия с другими регуляторными молекулами (рис. 1.105).
RUNX2 играет ведущую роль как в остеогенезе в период эмбрионального развития, так и в перестройке костной ткани во взрослом периоде, поэтому кодирующий его ген считают главным геном остеогенеза и развития кости (Mundlos, 1999). Исследования, проведенные на мышах, нокаутных по гену RUNX2, подтвердили эти данные: у гомозиготных мышей, у которых отсутствовали оба аллеля этого гена (RUNX2-/-) не наблюдалось каких-либо признаков оссификации. Они погибали вскоре после рождения из-за остановки дыхания. В отличие от них, у мышей, гетерозиготных по мутантному R UNX2, фенотипические скелетные проявления напоминали таковые у больных с ключично-черепным дизостозом (Komori et al., 1997; Otto et al., 1997).
Транскрипционный фактор RUNX2 регулирует экспрессию многих генов, влияющих на фенотип остеобластов, в частности экспрессию генов, кодирующих щелочную фосфатазу, коллаген I типа, коллагеназу 3 (матриксную металлопротеиназу-13), остеопонтин и остеокальцин. RUNX2 участвует в целом ряде процессов первичной и вторичной биоминерализации: дифференцировке костной и хрящевой ткани (Zelzer, Olsen, 2003), морфогенезе зубов (Aberg et al, 2004), появлении остеолитических очагов при раке молочной железы (Barnes et al., 2004), развитии артериосклероза (Cola et al., 2004), а также обызвествлении сосудистой стенки при хронической почечной недостаточности (Chen, Мое, 2004). RUNX2 влияет также на механобиологию фибробластов периодонта (Ziros et al., 2002) и остеобластов (Baumert et al., 2004).
После пионерской работы, опубликованной Mundlos и соавт. в 1997 г., в литературе было описано более 80 различных мутаций гена RUNX2 у больных с ключично-черепным дизостозом, затрагивающих отдельные нуклеотидные основания (точечные мутации) или более протяженные последовательности нуклеотидов.
Некоторые мутации негативно сказываются на распознавании последовательности ДНК или делают распознавание вовсе невозможным, либо дестабилизируют связь с ДНК; при других мутациях нарушается образование комплекса с субъединицей CBFβ. На рисунке 106 показано, как мутация может влиять на структуру и функцию транскрипционного фактора RUNX2. Мутации этого фактора приводят к развитию ключично-черепного дизостоза из-за недостаточности гена RUNX2 в гаплотипе: если сохраняется возможность экспрессии на гене «дикого» типа, например при гетерозиготной мутации, экспрессируется только 50% его продукта, чего бывает недостаточно для нормального развития и функции костной ткани. В этом смысле корреляция между генотипом и его фенотипическим проявлением в виде ключично-черепного дизостоза была бы, конечно, желательной. Однако трудно установить, имеется ли прямая корреляция между генотипом и фенотипом, так как данная аномалия встречается редко, а отчасти также из-за того, что ее клинические признаки, например сверхкомплектные зубы, проявляются только к определенному возрасту (Baumert et al., 2005, 2006).
Ключично-черепной дизостоз - наглядный пример того, насколько сложна зависимость фенотипических проявлений от генетических нарушений и как трудно установить генетический компонент. Дальнейшие исследования могут привести к модификации биомеханики перемещений зубов при тех или иных ортодонтических аномалиях развития.