Нарушения обмена аминокислот у детей: клиника, диагностика
Фенилкетонурия у детей. Распространённость фенилкетонурии в нашей стране составляет 1 на 10 000-15 000 живых новорождённых. Это связано или с дефицитом фермента фенилаланин-гидроксилазы (классическая фенилкетонурия), или с нарушением синтеза или распада его кофактора — биоптерина. Если не проводится лечение, то обычно наблюдается задержка в развитии начиная с 6-12-месячного возраста.
От детей может исходить заплесневелый запах метаболита фенилуксусной кислоты. Многие поражённые дети светловолосые и голубоглазые, у них развиваются экзема и судороги. К счастью, большинство детей с этим заболеванием выявляются по национальной программе биохимического скрининга (теста Гатри).
Лечение классической фенилкетонурии заключается в ограничении фенилаланина в диете до тех пор, пока этого достаточно для оптимального физического и неврологического развития. Регулярно производят мониторинг уровня фенилаланина в плазме. В настоящее время рекомендуется придерживаться этой диеты на протяжении всей жизни. Это особенно важно во время беременности, когда высокий уровень фенилаланина у матери может привести к повреждению плода.
Дефекты кофакторов, при которых прогноз намного хуже, чем при классической фенилкетонурии, лечатся уменьшением содержания фенилаланина в диете и введением предшественников нейромедиаторов.
Гомоцистеинурия у детей
Гомоцистеинурия у детей связана с дефицитом цистатионин-синтетазы. Проявляется задержкой развития и иногда подвывихом хрусталика глаза (эктопия хрусталика). Наблюдаются прогрессирующее затруднение в обучении, психические нарушения и эпилептические припадки. Скелетные проявления напоминают синдром Марфана. Цвет кожи обычно светлый, волосы ломкие. В любом возрасте могут происходить эпизоды тромбоэмболии. Практически половина больных детей хорошо реагирует на введение больших доз коэнзима пиридоксина.
Детям, не отвечающим на терапию, назначается диета с низким содержанием метионина, обогащенная цистеином и с добавлением реметилинизирующего агента бетаина.
Тирозинемия у детей
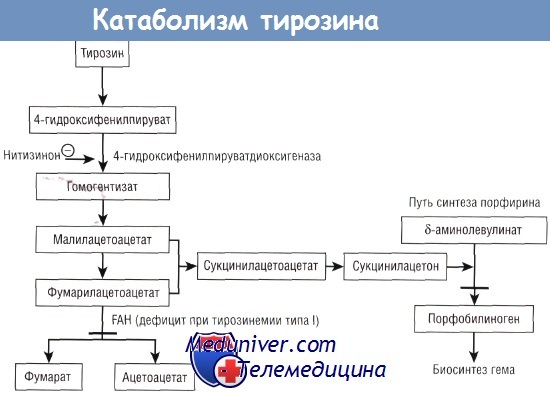
Тирозинемия (1-го типа) — редкое аутосомно-рецессивное заболевание, возникающее в результате дефицита фумарилацетоацетазы. Накопление токсических метаболитов вызывает повреждение печени (приводящее к печёночной недостаточности) и почечных канальцев (приводящее к синдрому Фанкони).
Если это заболевание не лечить, летальный исход неизбежен, однако в настоящее время доступна эффективная терапия препаратом, называемым «NTBC» (нитисинон, орфадин), который ингибирует ферменты, необходимые для катаболизма тирозина, наряду с диетой с пониженным содержанием тирозина и фенилаланина.
Нарушения обмена аминокислот проявляющиеся остро в неонатальном периоде:
• Нарушения катаболизма различных незаменимых аминокислот (аминокислот с разветвлёнными цепями, лейцина, изолейцина и валина, аминокислоты с нечётными цепями, например, треонина), которые приводят к формированию болезни кленового сиропа и другим нарушениям органических кислот.
• Дефекты цикла мочевины.
• Нарушения метаболизма углеводов — классическая галактоземия.
Тирозинемия типа I является следствием дефицита фумарилацетоацетат-гидролазы (FAH). Сукцинилацетон ингибирует синтез порфобилиногена. Нитизинон ингибирует фермент 4-гидроксифенилпируватдиоксигеназу.
При этих заболеваниях поражённый ребёнок при рождении выглядит нормально, но через несколько дней развиваются неспецифические признаки и симптомы, схожие с симптомами других, более распространённых заболеваний, таких как генерализованная инфекция. Наиболее распространённые признаки заболевания.
• Рвота, ацидоз и нарушение кровообращения с последующим нарушением сознания и судорогами — заставляют предполагать одну из органических ацидемий.
• Неврологические признаки летаргии, отказ от приёма пищи, гипотония, сонливость, потеря сознания и апноэ — предполагают первичные дефекты цикла мочевины. При внутривенной инфузии жидкостей наступает улучшение, однако рецидив при возобновлении молочного питания является характерным признаком классической галактоземии.
Диагноз устанавливается на основании «метаболического скрининга» в дополнение к стандартным исследованиям младенцев в тяжёлом состоянии. «Метаболический скрининг» варьирует в разных лабораториях, и его необходимо обсудить с сотрудниками лаборатории до сбора образцов. Также должно быть показано неотложное проведение исследования. Вероятнее всего, потребуются образцы крови и мочи. Простой тест у больничной койки на кетоны может быть полезным, поскольку при тяжёлом кетоацидозе и ацидозе у младенца с энцефалопатией есть веские основания предполагать нарушения метаболизма органических кислот.
У пациентов с ацидозом необходимо вычислить анионную разницу (сумма плазменных концентраций натрия и калия минус сумма концентраций хлоридов и бикарбонатов), что может быть полезным. Значения выше 25 ммоль/л (в норме — 12-16 ммоль/л) обычно развиваются вторично по отношению к органической ацидемий. Хорошей практикой является сбор мочи, выделяемой у младенца для возможного анализа в будущем (или до тех пор, пока диагноз не установлен), равно как и сбор образцов крови до проведения трансфузии любых компонентов крови, если последние противоречат интерпретации лабораторных тестов.
Как краткосрочное, так и долговременное ведение пациентов зависит от основного диагноза. В неотложной экстренной ситуации ликвидация токсических метаболитов и ограничение катаболизма имеют наивысшее значение. Часто требуются перевод в отделение интенсивной терапии новорождённых, механическая вентиляция и гемодиализ. Долговременное ведение пациентов включает профессиональную коррекцию диеты, а также использование специальных лекарственных препаратов в зависимости от основного диагноза.