а) Неонатальный холестаз. Неонатальный холестаз биохимически определяется как устойчивое повышение уровня конъюгированного билирубина в сыворотке крови ребенка >14 дней жизни. В тех случаях, когда желтуха впервые появляется в возрасте >14 дней жизни, не разрешается к этому сроку или продолжает прогрессировать, необходима клиническая оценка и определение уровня конъюгированного билирубина.
Причинами холестаза у новорожденных м.б. инфекционные, генетические, метаболические аномалии, также он может возникать по невыясненным причинам. Все вышеперечисленное может привести к механической обструкции ЖВП или к функциональным нарушениям экскреторной функции печени и секреции желчи (табл. 1). К механическим факторам относятся стриктуры и обструкция общего желчного протока. Билиарная атрезия — наиболее типичная обструктивная аномалия. Функциональные нарушения секреции желчи м.б. обусловлены врожденными дефектами, повреждением клеток печени или аппарата желчной секреции.
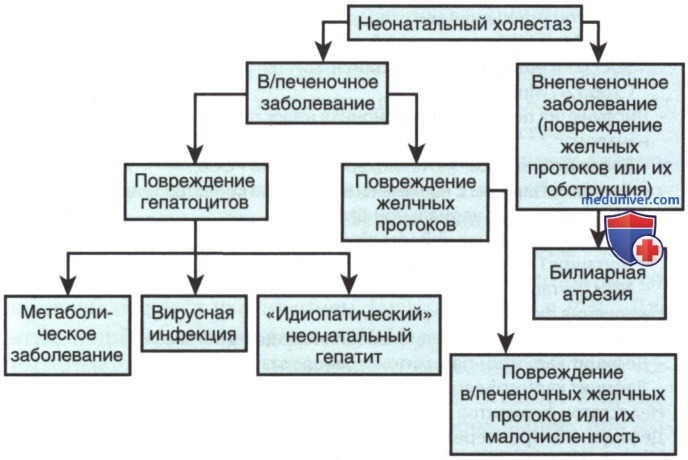
Неонатальный холестаз можно разделить на внепеченочный и в/печеночный (рис. 1). Клиника различных форм холестаза остается схожей. Провести диагностику некоторых отдельных заболеваний, таких как галактоземия, муковисцидоз, сепсис или гипотиреоз у новорожденных относительно легко, так как эти заболевания входят в большинство программ неонатального скрининга. Но во многих случаях причина холестаза не столь ясна. Особенно сложно дифференцировать билиарную атрезию и идиопатический неонатальный гепатит.
Рисунок 1. Неонатальный холестаз. Концептуальный подход к группе заболеваний, проявляющихся неонатальным холестазом. Некоторые заболевания пересекаются. У пациентов с билиарной атрезией может наблюдаться некоторая степень в/печеночного повреждения. У пациентов с «идиопатическим» неонатальным гепатитом в более позднем возрасте могут определяться первичные метаболические или вирусные заболевания
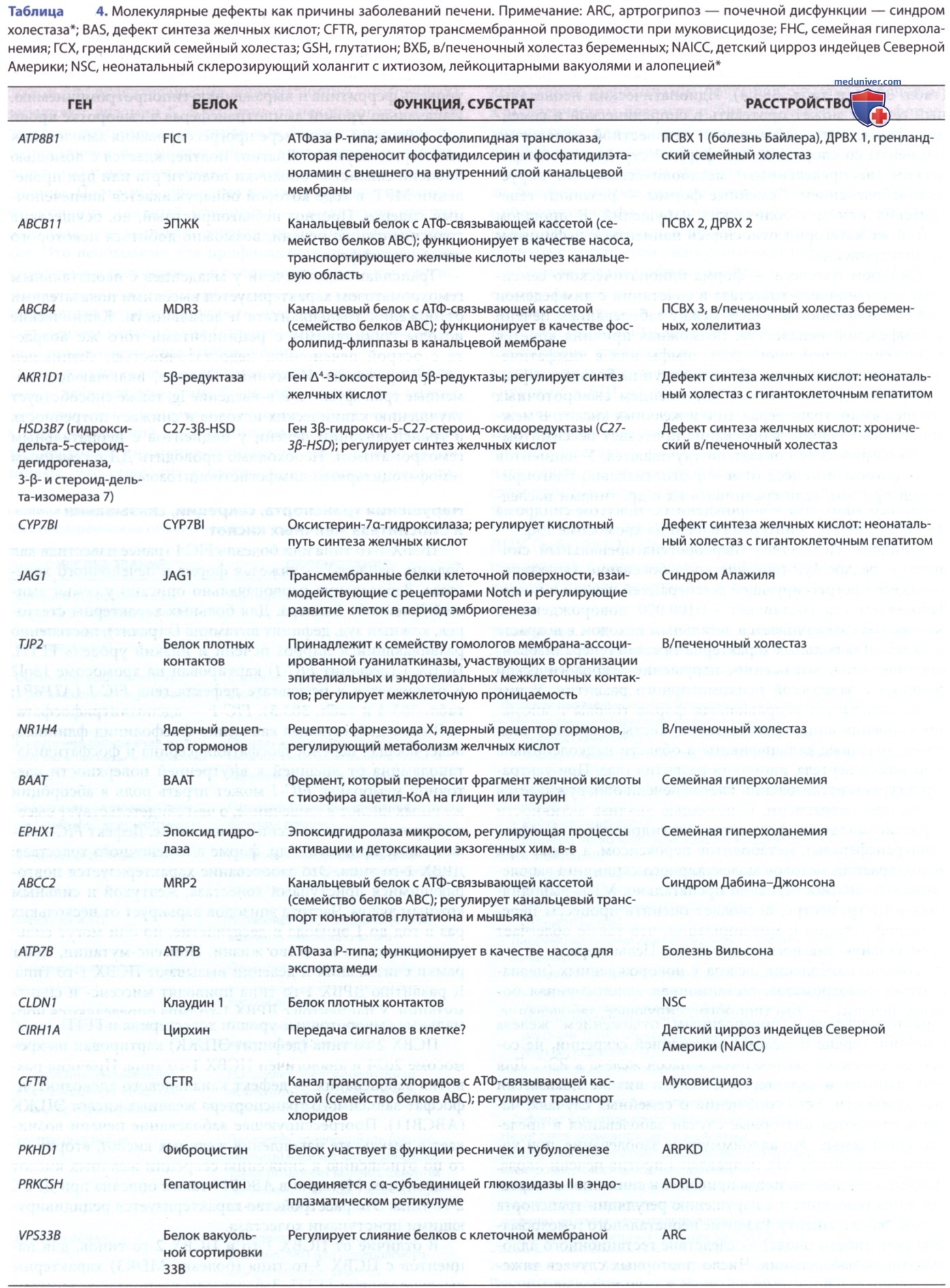
1. Механизмы. Метаболическое заболевание печени, вызванное врожденными нарушениями синтеза или транспорта желчных кислот, сочетается с накоплением атипичных токсичных желчных кислот и неспособностью продуцировать нормальные холеретические и трофические желчные кислоты. Клинические и гистологические проявления неспецифичны, сходны с др. формами гепатобилиарных повреждений у новорожденных. Некоторые неясные формы поражений печени у новорожденных вызваны аутоиммунными процессами.
Некоторые гистологические варианты повреждения печени, характерные для детей раннего возраста, не наблюдаются у пациентов старшего возраста. Гигантоклеточная трансформация гепатоцитов обнаруживается у младенцев с существующим холестазом. Она может возникать при любой форме неонатального повреждения печени, но чаще всего встречается и тяжелее протекает при в/печеночных формах холестаза. Клинические и гистологические данные от пациентов с неонатальным гепатитом и билиарной атрезией схожи, но в каждом случае есть свои отличительные особенности. Главное сходство этих двух патологий — неясный источник исходного повреждения, приводящего к развитию воспаления клеток печени или ЖВП. Если основной очаг поражения — эпителий желчных протоков, может развиться холангит. Он приводит к прогрессирующему склерозу и сужению желчного дерева, вплоть до его полной облитерации (билиарная атрезия).
Повреждение клеток печени может приводить к развитию клинической и гистологической картины «неонатального гепатита». Эта концепция не разъясняет точный механизм, но хорошо объясняет документированные случаи неожиданного постнатального развития заболевания. Напр., когда у младенцев с функционирующей (по данным холангиографии) билиарной системой, которым изначально был установлен диагноз неонатального гепатита, в дальнейшем развивалась билиарная атрезия. Также развитие холестаза у новорожденных м.б. обусловлено функциональными нарушениями образования желчи. Процесс оттока желчи напрямую зависит от эффективности экскреции желчных кислот гепатоцитами.
В раннем возрасте, в период относительно неэффективного метаболизма гепатоцитов и транспорта желчных кислот, даже незначительные повреждения печени могут усугубить нарушения оттока желчи. Это приводит к выработке атипичных и потенциально токсичных желчных кислот. Даже единичное нарушение на одном из этапов процесса печеночной экскреции приводит к развернутому холестатическому синдрому. У младенцев с различными формами в/печеночного холестаза обнаруживаются специфические дефекты синтеза желчных кислот (см. табл. 1). Тяжелые формы семейного холестаза ассоциируются с неонатальным гемохроматозом, гестационной аллоиммунной болезнью печени (реакция материнские АТл против гепатоцитов плода), которое реагирует на в/в-Ig, вводимый матери. Также известны случаи холестаза при сепсисе, предположительно, обусловленные влиянием эндотоксина Escherichia coli.
2. Оценка. При выявлении холестаза необходимо определить его точную причину (табл. 2). Холестаз у новорожденного м.б. начальным проявлением целого ряда потенциально опасных заболеваний со схожими проявлениями, поэтому клиника дает мало информации об этиологии. Желтуха, потемнение мочи, светлый или ахоличный стул и гепатомегалия у больного ребенка — следствие нарушения оттока желчи, результат повреждения гепатоцитов или обструкции ЖВП. Нарушение синтетической функции печени может привести к гипопротромбинемии и кровотечениям. В схему первичной терапии младенцев с холестазом для предотвращения кровоизлияний (в/черепных) добавляют витамин К. В отличие от неконъюгированной гипербилирубинемии, которая м.б. физиологической, холестаз (любое повышение уровня конъюгированного билирубина) у новорожденных — всегда патологическое состояние. Поэтому необходимо незамедлительно выяснять его причину. Главный приоритет — выявление причин холестаза, для которых существует специфическое лечение.
Это необходимо для профилактики дальнейшего повреждения и долгосрочных осложнений, таких как сепсис, эндокринопатия (гипотиреоз, пангипопитуитаризм), нутриционная гепатотоксичность, вызываемая конкретным нарушением метаболизма (галактоземия), и прочие метаболические нарушения (тирозинемия).
Др. потенциально излечимое метаболическое заболевание, дефицит лизосомной кислой липазы. Это редкая АуР-лизосомная болезнь накопления, результат мутации в гене лизосомной кислой липазы (LIPA). Эта мутация обусловливает снижение активности лизосомной кислой липазы. Это приводит к накоплению эфиров холестерина и в меньшей степени триглицеридов во многих органах и тканях, включая печень, селезенку, надпочечники, ЛУ, слизистую оболочку кишечника, эндотелий сосудов и скелетные мышцы. Клинически заболевание представлено двумя фенотипами: болезнь Вольмана с началом в младенческом возрасте и болезнь накопления эфиров холестерина, для которой характерно более позднее начало. Дефицит лизосомной кислой липазы впервые проявляется в младенческом возрасте, протекает остро и тяжело, быстро прогрессируя до печеночной недостаточности. Себелипаза альфа (рекомбинантный человеческий фермент лизосомная кислая липаза) одобрена для лечения пациентов с дефицитом лизосомной кислой липазы.
Поражение гепатобилиарной системы может выступать в качестве начального проявления гомозиготного фенотипа недостаточности α1-антитрипсина и муковисцидоза. Развитие заболевания печени в неонатальном периоде м.б. обусловлено врожденным сифилисом, а также специфическими вирусными инфекциями, в особенности ЕСНО-вирусами и ВПГ, включая ЦМВ. На их долю приходится небольшой процент случаев синдрома неонатального гепатита. Вирусы гепатита (А, В, С) крайне редко бывают причиной холестаза у новорожденных. Митохондриальные нарушения могут проявляться острой печеночной недостаточностью или холестазом в неонатальном периоде. Самые частые среди таких нарушений — дефекты дыхательной цепи и синдромы истощения митохондриальной ДНК (табл. 3). Последний важный шаг в оценке новорожденных с холестазом — дифференцировать внепеченочную билиарную атрезию и неонатальный гепатит.
3. Внутрипеченочный холестаз:
- Неонатальный гепатит. Под неонатальным гепатитом подразумевается в/печеночный холестаз (рис. 1) в различных его формах (табл. 1 и табл. 4). Идиопатический неонатальный гепатит может протекать в спорадической и семейной форме. Это заболевание неизвестной этиологии. Пациенты со спорадической формой страдают специфическим (неопределенным) метаболическим или вирусным заболеванием. Семейные формы — результат генетических или метаболических нарушений. В прошлом к этой же категории относились пациенты с дефицитом α1-антитрипсина.
Синдром Аагенеса — форма идиопатического семейного в/печеночного холестаза в сочетании с лимфедемой нижних конечностей. Связь между заболеванием печени и лимфедемой неизвестна. Возможная причина кроется в снижении печеночного тока лимфы или в лимфатической гипоплазии печени. Для этой группы больных характерны эпизоды холестаза с повышением сывороточных уровней аминотрансфераз, ЩФ и желчных кислот. В межприступный период заболевание протекает бессимптомно, биохимические показатели улучшаются. У пациентов с синдромом Аагенеса отмечен относительно благоприятный прогноз, если сравнивать их с др. типами наследственного холестаза новорожденных. Локусом синдрома Аагенеса считается интервал 6.6 сМ на хромосоме 15q.
Синдром Цельвегера (цереброгепаторенальный синдром) — редкое АуР-генетическое заболевание, характеризующееся прогрессирующей дегенерацией печени и почек. Заболеваемость составляет ~1:100 000 новорожденных. Заболевание заканчивается летальным исходом в возрасте 6-12 мес. Для больных характерна тяжелая генерализованная гипотония, выраженное нарушение неврологической функции с задержкой психомоторного развития. У них также отмечается неправильная форма головы и аномалии строения лица, гепатомегалия, кисты коркового слоя почек, точечные кальцификаты в области надколенников и большого вертела, аномалии развития глаз. При ультраструктурном исследовании клеток печени обнаруживается отсутствие пероксисом.
С помощью анализа активности пероксисомальных ферментов (дигидроацетон-фосфатацилтрансферазы), метаболитов пероксисом, а также при использовании методов молекулярного скрининга заболевание м.б. диагностировано пренатально. МРТ, выполняемая в III триместре, позволяет оценить процессы церебральной гирации и миелинизации, что также облегчает пренатальную диагностику синдрома Цельвегера.
Болезнь накопления железа у новорожденных (неонатальный гемохроматоз, гестационная аллоиммунная болезнь печени) — быстропрогрессирующее заболевание, характеризующееся повышенным отложением железа в печени, сердце и железах внутренней секреции, не сопровождающееся увеличением запасов железа в РЭС. Для этих пациентов характерен СПОН и низкие показатели выживаемости. Есть сообщения о семейных случаях, часто встречаются повторные случаи заболевания в пределах одной семьи. Это аллоиммунное заболевание, при котором материнские АТл направлены против печени плода. Повреждение печени плода приводит к снижению экспрессии в ней гепсидина и к нарушению регуляции транспорта железа через плаценту. Развитие неонатального гемохроматоза (или гибели плода) — следствие гестационного аллоиммунного заболевания. Число повторных случаев тяжелого неонатального гемохроматоза в группе повышенного риска м.б. уменьшено путем еженедельного введения матери высоких доз (1 г/кг) в/в Ig с 18-й неделе беременности.
Лабораторные данные включают гипогликемию, гипербилирубинемию, гипоальбуминемию, повышенный уровень ферритина и выраженную гипопротромбинемию. Изначально уровни аминотрансфераз в сыворотке крови м.б. высокими, но по мере прогрессирования заболевания они нормализуются. Диагноз подтверждается с помощью биопсии слизистой оболочки полости рта или при проведении МРТ, в ходе которой обнаруживается внепеченочный сидероз. Прогноз неблагоприятный, но, осуществив трансплантацию печени, возможно добиться некоторого лечебного эффекта.
Трансплантация печени у младенцев с неонатальным гемохроматозом характеризуется высокими показателями отторжения трансплантата и летальности. Клинические исходы сопоставимы с реципиентами того же возраста с острой печеночной недостаточностью, возникшей по др. причинам. Иммунная терапия, включающая обменные трансфузии и в/в-введение Ig, также способствует улучшению клинических исходов и снижает потребность в трансплантации печени у пациентов с неонатальным гемохроматозом. Необходимо проводить ДД с семейным гемофагоцитарным лимфогистиоцитозом.
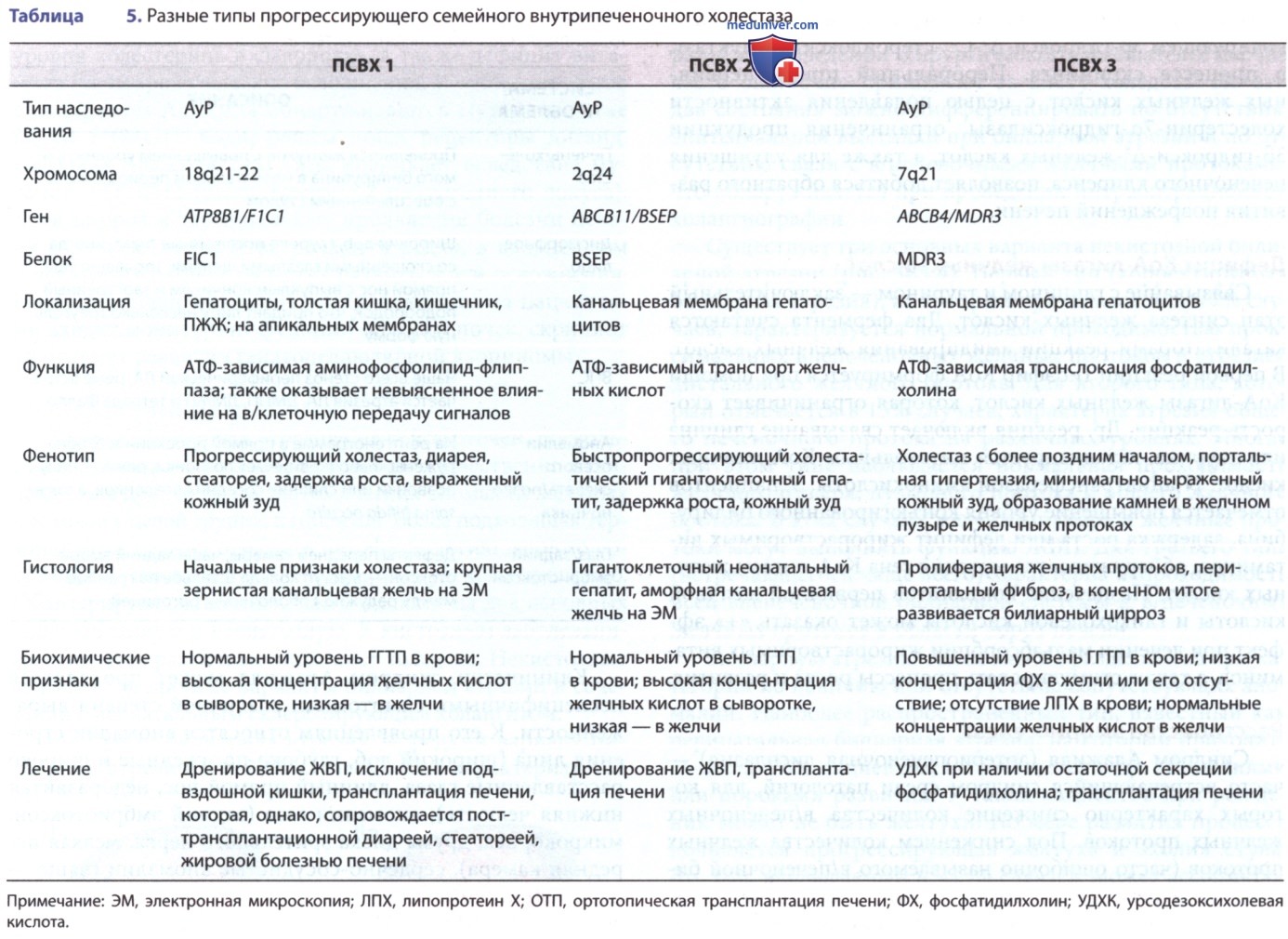
- Нарушения транспорта, секреции, связывания и биосинтеза желчных кислот. ПСВХ 1-го типа или болезнь FIC 1 (ранее известная как болезнь Байлера) — тяжелая форма в/печеночного холестаза. Заболевание первоначально описано у семьи амишей Джейкоба Байлера. Для больных характерны стеаторея, кожный зуд, дефицит витамина D (рахит), постепенно развивающийся цирроз печени и низкий уровень ГГТП. ПСВХ 1 (дефицит FIC-1) картирован на хромосоме 18ql2 и развивается в результате дефекта гена FIC-1 (АТР8В1; табл. 4 и табл. 5). FIC-1 — аденозинтрифосфатаза P-типа, действующая как амино-фосфолипид-флиппаза, облегчающая перенос фосфатидилсерина и фосфатидилэтаноламина от внешней к внутренней поверхности клеточной мембраны. FIC-1 может играть роль в абсорбции желчных кислот в кишечнике, о чем свидетельствует высокий уровень ее экспрессии в кишечнике.
Дефект FIC-1 может также привести к др. форме в/печеночного холестаза: ДРВХ 1-го типа. Это заболевание характеризуется повторяющимися приступами холестаза, желтухой и сильным кожным зудом. Частота эпизодов варьирует от нескольких раз в год до 1 эпизода в десятилетие, но они могут сильно повлиять на качество жизни. Нонсенс-мутации, сдвиг рамки считывания и делеции вызывают ПСВХ 1-го типа. К развитию ДРВХ 1-го типа приводят миссенс- и сплит-мутации. У пациентов с ДРВХ 1-го типа определяются нормальные сывороточные уровни холестерина и ГГТП.
ПСВХ 2-го типа (дефицит ЭПЖК) картирован на хромосоме 2q24 и аналогичен ПСВХ 1-го типа. Причина развития заболевания — дефект канальцевого аденозинтрифосфат-зависимого транспортера желчных кислот ЭПЖК (АВСВ11). Прогрессирующее заболевание печени возникает в результате накопления желчных кислот, вторичного по отношению к снижению секреции желчных кислот в канальцах. Мутация в АВСВ11 также описана при ДРВХ 2-го типа. Это расстройство характеризуется рецидивирующими приступами холестаза.
В отличие от ПСВХ 1-го и ПСВХ 2-го типов, для пациентов с ПСВХ 3-го типа (болезнь MDR3) характерны высокие уровни ГГТП. Заболевание возникает вследствие дефекта канальцевой фосфолипидной флиппазы, MDR3 (АВСВ4). Это приводит к недостаточной транслокации фосфатидилхолина через канальцевую мембрану. У матерей, гетерозиготных по этому гену, во время беременности может развиться в/печеночный холестаз.
Семейная гиперхоланемия характеризуется повышенной концентрацией желчных кислот в сыворотке крови, кожным зудом, задержками в развитии и коагулопатией. Семейная гиперхоланемия — сложный генетический дефект, в основе которого лежат мутации коэнзима А желчных кислот (КоА), N-ацилтрансферазы аминокислот (кодируемой ВААТ), а также мутации белка плотных контактов 2 (кодируемого TJP 2, также известным как ZO-2). Мутация в ВААТ, ферменте, ответственном за связывание желчных кислот, полностью нивелирует его активность. У пациентов, гомозиготных по данной мутации, в составе желчи содержатся только неконъюгированные желчные кислоты. В результате мутации ВААТ и TJP 2 может нарушаться транспорт и циркуляция желчных кислот. Пациенты с семейной гиперхоланемией чаще хорошо реагируют на урсодезоксихолевую кислоту.
Триггерный или поддерживающий фактор развития холестаза у новорожденных — дефекты биосинтеза желчных кислот. Эти дефекты приводят к отсутствию у ребенка нормальных первичных трофических или холеретических желчных кислот, накоплению их атипичных (гепатотоксичных) метаболитов. Врожденные нарушения синтеза желчных кислот приводят к острым и хроническим заболеваниям печени. Ранняя диагностика позволяет своевременно назначить необходимую заместительную терапию, которая поможет обратить вспять развивающееся повреждение печени. Существуют описания нескольких конкретных дефектов.
Дефицит Δ4-3-оксостероид-5β редуктазы, четвертой ступени на пути деградации холестерина до первичных желчных кислот, проявляется выраженным холестазом и печеночной недостаточностью, развивающимися вскоре после рождения, коагулопатией и метаболическим повреждением печени, напоминающим тирозинемию. Гистологически печень характеризуется нарушением дольковой структуры с наличием гигантских клеток, псевдоацинарной трансформацией и канальцевым застоем желчи. При проведении масс-спектрометрии обнаруживается повышенная экскреция желчных кислот с мочой и преобладание оксогидрокси- и оксо-дигидроксихоленовых кислот. Диагноз м.б. установлен путем скринингового выявления мутаций в SRD5B1 (AKR1D1). Этот ген кодирует Δ4-3-оксостероид 5β-редуктазу. Назначение холевой и урсодезоксихолевой кислот позволяет достичь нормализации биохимических, гистологических и клинических показателей.
Дефицит 3β-гидрокси-Δ5-С27-стероидоксидоредуктазы, второй ступени синтеза желчных кислот из холестерина, обусловливает развитие ПСВХ. У пациентов наблюдается желтуха с повышенным уровнем аминотрансфераз и гепатомегалия. При этом сывороточные уровни ГГТП и холилглицина остаются нормальными. Гистологическая картина варьирует от гигантоклеточного до хронического гепатита. Диагноз можно заподозрить при обнаружении в моче методом масс-спектрометрии С24-желчных кислот, сохраняющих структуру 3β-гидрокси-Δ5. Окончательно диагноз подтверждается обнаружением мутаций в гене HSD3B7, кодирующем 3β-гидрокси-Δ5-С27-стероидоксидоредуктазу, в процессе скрининга. Пероральный прием первичных желчных кислот с целью подавления активности холестерин-7α-гидроксилазы, ограничения продукции 3β-гидрокси-Δ5-желчных кислот, а также для улучшения печеночного клиренса, позволяет добиться обратного развития повреждений печени.
4. Дефицит КоА-лигазы желчных кислот. Связывание с глицином и таурином — заключительный этап синтеза желчных кислот. Два фермента считаются катализаторами реакции амидирования желчных кислот. В первой реакции тиоэфир КоА формируется при помощи КоА-лигазы желчных кислот, которая ограничивает скорость реакции. Др. реакция включает связывание глицина или таурина, катализируется цитозольным КоА желчных кислот: N-ацилтрансферазой аминокислоты. У пациентов отмечается повышение уровня конъюгированного билирубина, задержка роста или дефицит жирорастворимых витаминов, обнаруживается мутация гена КоА-лигазы желчных кислот. Назначение конъюгатов первичной желчной кислоты и гликохолевой кислоты может оказать «+» эффект при лечении мальабсорбции жирорастворимых витаминов, а также стимулировать процессы роста и развития.
5. Расстройства эмбриогенеза. Синдром Алажиля (артериопеченочная дисплазия) — часто встречающийся синдром среди патологий, для которых характерно снижение количества в/печеночных желчных протоков. Под снижением количества желчных протоков (часто ошибочно называемого в/печеночной билиарной атрезией) понимают отсутствие или заметное снижение числа междольковых желчных протоков в портальных триадах. При этом сохраняется нормальный размер ветвей воротной вены и печеночных артериол. При проведении биопсии в раннем возрасте обнаруживается воспалительный процесс с вовлечением желчных протоков. В дальнейшем воспалительные процессы становятся менее активными, но отмечается остаточное снижение количества желчных протоков и их диаметра. Это может напоминать картину, характерную для синдрома исчезновения желчных протоков, который наблюдается у взрослых людей с иммуноопосредованными заболеваниями. При последовательных гистологических исследованиях печени часто наблюдается картина прогрессирующей деструкции желчных протоков.
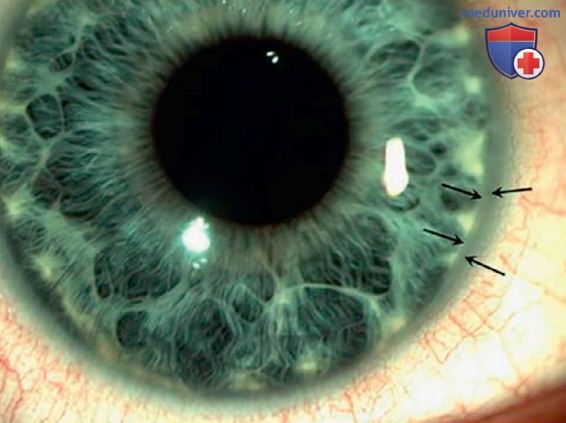
Клинически синдром Алажиля может проявляться неспецифичными симптомами различной степени выраженности. К его проявлениям относятся аномалии строения лица (широкий лоб, глубоко посаженные и широко расставленные глаза, длинный прямой нос, недоразвитая нижняя челюсть), аномалии глаз (задний эмбриотоксон, микрокорнеа, друзы диска зрительного нерва, мелкая передняя камера), сердечно-сосудистые аномалии (чаще — периферический стеноз ЛА, реже — тетрада Фалло, атрезия ЛА, ДМЖП и ДМПП, коарктация аорты), дефекты позвонков (бабочковидные позвонки, сросшиеся позвонки, spina bifida occulta, аномалии ребер) и тубулоинтерстициальная нефропатия (табл. 6, рис. 2-3). Др. признаки, такие как низкий рост, недостаточность ПЖЖ, васкулопатия (синдром Моямоя, инсульт), нарушения сперматогенеза, м.б. следствием дефицита нутриентов или его причиной. У пациентов с синдромом Алажиля при отсутствии лечения может развиваться кожный зуд и ксантомы в сочетании с выраженным повышением уровня холестерина в сыворотке, а также дефицит витамина Е и неврологические осложнения.
Рисунок 2. Задний эмбриотоксон
Рисунок 3. Бабочковидные позвонки в грудном и верхнем поясничном отделах. Ребенок перенес операцию на сердце, поэтому на изображении можно увидеть проволочную сетку
У 90% пациентов с синдромом Алажиля обнаруживаются мутации в гене Jagged 1 (JAG1), кодирующем notch рецепторы лиганд. Синдром Алажиля 2-го типа возникает вследствие мутаций NOTCH2 (белок-гомолог 2 нейрогенного локуса). Хотя цирроз и терминальные проявления болезни печени в раннем возрасте встречаются редко, в дальнейшем у некоторых пациентов могут развиться эти осложнения. Поэтому в план долгосрочного наблюдения за пациентами входит мониторинг функции сердца и почек, скрининг на предмет развития гепатоцеллюлярной карциномы.
6. Билиарная атрезия. Термин «билиарная атрезия» не совсем точен, поскольку он охватывает большое количество существенно отличающихся друг от друга анатомических аномалий желчных протоков у целой группы пациентов. Более подходящая терминология должна отражать патофизиологию данного состояния — некистозную облитерирующую холангиопатию. Облитерирующая холангиопатия делится на два основных типа: кистозную и некистозную. К кистозным заболеваниям относятся различные типы кист холедоха. Некистозные формы — различные варианты билиарной атрезии в сочетании с неонатальным склерозирующим холангитом.
Кистозная билиарная атрезия — редкий вариант билиарной атрезии (10-20% случаев). Она характеризуется относительно благоприятным прогнозом, особенно при раннем проведении хирургического вмешательства. Ее часто ошибочно принимают за кисту холедоха. Но эти два состояния можно дифференцировать по отсутствию эпителиальной выстилки при билиарной атрезии и по отсутствию связи с в/печеночными желчными протоками, что обнаруживается при проведении интраоперационной холангиографии.
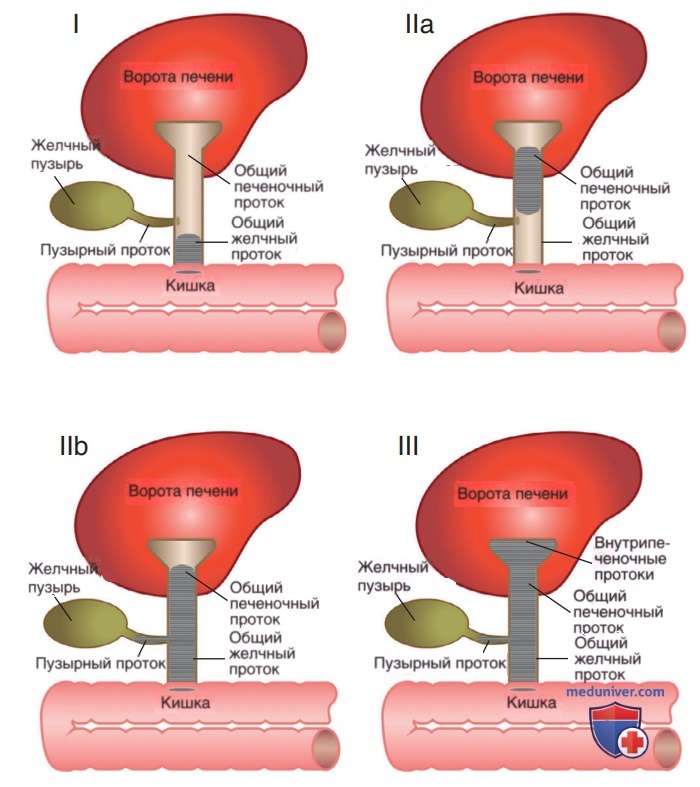
Существует три основных варианта некистозной билиарной атрезии (рис. 4). Первый тип (корректируемая билиарная атрезия), на который приходится лишь 7% случаев, характеризуется нормальной проходимостью проксимальных внепеченочных желчных протоков и атрезией дистального желчного протока. Для второго типа, который отмечается в 15% случаев, характерна атрезия общего печеночного протока на различных уровнях. Иногда при этом типе наблюдается нормальная проходимость желчного пузыря, пузырного протока и общего желчного протока. В этих случаях желчный пузырь и желчные протоки могут выполнять функцию ЖВП. Для третьего типа (встречающегося чаще всего) характерна непроходимость всей внепеченочной билиарной системы и в/печеночных желчных протоков в области ворот печени.
Рисунок 4. Классификация билиарной атрезии в зависимости от области поражения (серый цвет). Тип I: атрезия дистального желчного протока в сочетании с проходимым проксимальным внепеченочным желчным протоком. Тип IIа: атрезия общего печеночного протока. Тип IIb: атрезия общего печеночного протока, пузырного протока и общего желчного протока. Тип III: непроходимость всей внепеченочной билиарной системы, а также в/печеночных желчных протоков в области ворот печени
Билиарную атрезию также можно разделить на три категории по наличию или отсутствию сопутствующих аномалий. Наиболее распространенный тип, известный как перинатальная билиарная атрезия, на который приходится около 70% пациентов, не сочетается с др. аномалиями или пороками развития. У таких пациентов при рождении может не быть желтухи. По мере развития процесса появляется прогрессирующая желтуха и ахолия стула. Др. тип, наблюдаемый в ~15% случаев, может ассоциироваться с гетеротаксией (включая обратное расположение внутренних органов — situs inversus), мальротацией, полиспленией, перерывом НПВ, а также с ВПС. Этот тип также известен как синдром билиарной атрезии и мальформаций селезенки, и чаще всего он характеризуется неблагоприятным прогнозом. При третьем типе, в который входят оставшиеся 15% случаев, могут обнаруживаться и др. врожденные пороки развития, напр. кисты холедоха, аномалии почек и ВПС.
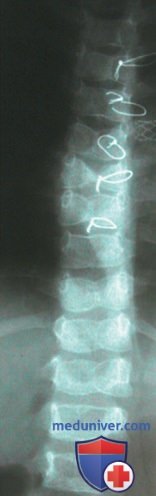
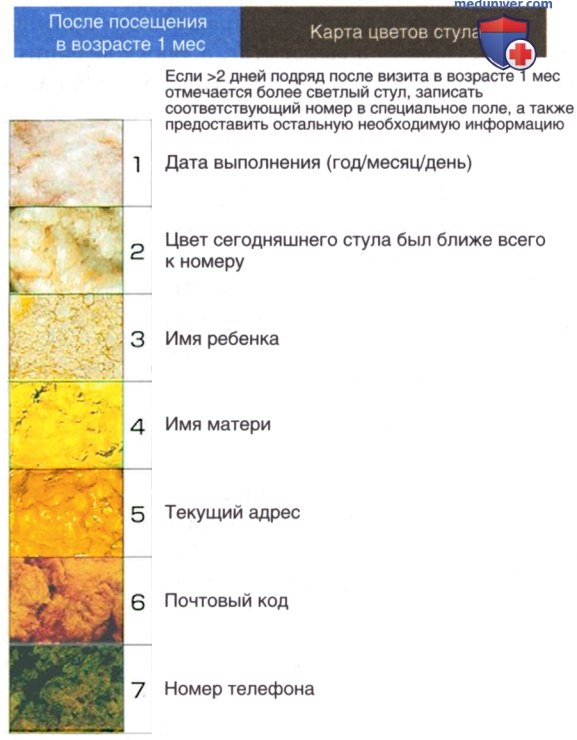
Частота билиарной атрезии составляет 1:10 000-15 000 новорожденных. Билиарная атрезия чаще встречается в странах Восточной Азии. Патология наблюдается у доношенных и недоношенных детей. Скрининг на предмет билиарной атрезии проводится не всем новорожденным. С некоторым успехом применяются карты эталонных цветов стула, помогающие обнаружить его ахолию (рис. 5). Любому младенцу >2 нед с впервые возникшей или персистирующей желтухой необходимо определять уровни общего и конъюгированного билирубина для выявления холестаза.
Рисунок 5. Пример карты цветов стула, которая использовалась в префектуре Тотиги с августа 1994 г. по март 2011 г. Карта представляла собой семь фотографий стула здоровых младенцев и младенцев с билиарной атрезией, имеющего различную окраску. На 1-3 изображениях запечатлен стул аномальной окраски, а на 4-7 — стул нормального цвета.
- Дифференциальная диагностика идиопатического неонатального гепатита и билиарной атрезии. Иногда трудно дифференцировать детей с билиарной атрезией, нуждающихся в хирургической помощи, от детей с в/печеночным заболеванием (неонатальным гепатитом) и проходимыми желчными протоками. Ни один биохимический тест или визуализационное исследование не дают достаточно информации. В алгоритмы диагностики входит анализ клинических, анамнестических, биохимических и радиологических данных.
Частота семейных случаев идиопатического неонатального гепатита составляет 20%, а для билиарной атрезии семейные случаи не характерны. Лишь у небольшой части младенцев с фетальным началом билиарной атрезии отмечается повышенная частота сопутствующих аномалий, напр. синдрома полисплении с гетеротаксией органов БП, мальротации, левокардии и интраабдоминальных сосудистых аномалий. Стойкая ахолия стула указывает на обструкцию ЖВП (атрезию), но и у пациентов с тяжелым идиопатическим неонатальным гепатитом могут наблюдаться транзиторные тяжелые нарушения экскреции желчи. Стабильная пигментация стула свидетельствует против билиарной атрезии. При пальпации печени у пациентов с билиарной атрезией могут обнаруживаться аномальные размеры или консистенция печени, что при идиопатическом неонатальном гепатите встречается гораздо реже.
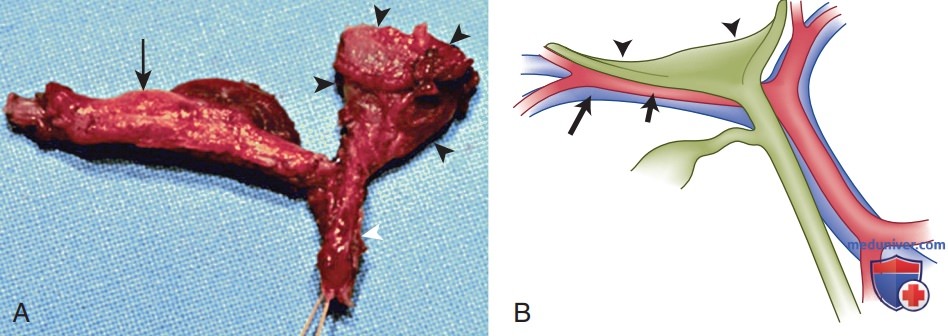
УЗИ ОБП — полезный метод диагностики при оценке неонатального холестаза. УЗИ позволяет выявить холедохолитиаз, перфорацию желчного протока и прочие структурные аномалии ЖВП, напр. кисты холедоха. У пациентов с билиарной атрезией при УЗИ можно обнаружить сопутствующие аномалии (абдоминальную полисплению, сосудистые мальформации). Также у пациентов с билиарной атрезией желчный пузырь не визуализируется совсем либо имеет очень малые размеры. Аналогичные результаты УЗИ могут наблюдаться у детей с в/печеночным холестазом, обусловленным идиопатическим неонатальным гепатитом, муковисцидозом или полным парентеральным питанием. Также для пациентов с билиарной атрезией характерен ультрасонографический симптом треугольного рубца — наличие конусообразной фиброзной массы, которая располагается краниальнее бифуркации воротной вены (рис. 6 и 7). Повышенная эхогенная плотность в области ворот печени отражает фиброзные изменения, характерные для билиарной атрезии.
Рисунок 6. Хирургические находки при билиарной атрезии: А — фотография операционного препарата облитерированных внепеченочных желчных протоков, показывающих фиброзный остаток протока (черные стрелки) в воротах печени, атрезию желчного пузыря (стрелка), а также фиброзированный общий желчный проток (белая стрелка). Фиброзный остаток протока — образование треугольно-конусовидной формы; В — схематическое изображение анатомических взаимоотношений между фиброзным остатком протока и кровеносными сосудами, окружающими ворота печени. Треугольный конусообразный фиброзный остаток протока (отмечен черными стрелками, зеленый цвет) располагается немного выше и спереди от воротной вены (длинная стрелка, синий цвет) и печеночной артерии (короткая стрелка, красный цвета)
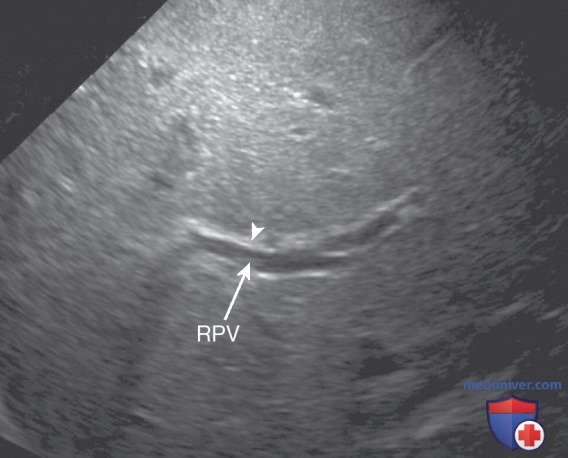
Рисунок 7. Билиарная атрезия у мальчика 8 нед с повышенным уровнем конъюгированного билирубина. На поперечной эхограмме виден симптом треугольного рубца, который представляет собой линейный тяж повышенной эхогенности (отмечен стрелкой) вдоль правой ветви воротной вены (RPV)
Повышенная эхогенная плотность в области ворот печени также полезна при оценке пациентов с неонатальным холестазом. Высокочастотная ультрасонография позволяет добиться лучшего разрешения за счет уменьшения глубины проникновения волны. Этот метод диагностики билиарной атрезии более эффективен по сравнению с обычным УЗИ.
Сцинтиграфия гепатобилиарной системы с меченными технецием производными иминодиуксусной кислоты — чувствительное исследование, не обладающее высокой специфичностью в отношении билиарной атрезии. С его помощью невозможно обнаружить др. структурные аномалии желчного дерева или сосудистые аномалии. Низкая специфичность и длительная подготовка (5 сут предварительной нагрузки фенобарбиталом) делают это исследование непрактичным и ограничивают его применение при обследовании детей с подозрением на билиарную атрезию.
Роль эндоскопической РХПГ и магнитно-резонансной РХПГ в диагностике билиарной атрезии не определена.
Чрескожная биопсия печени — ценное исследование в диагностике неонатальных заболеваний гепатобилиарной системы. Чрескожная биопсия печени позволяет обнаружить наиболее надежные отличительные признаки патологий. Для билиарной атрезии характерна пролиферация желчных протоков, желчные пробки, портальный или перилобулярный отек, фиброз при сохранении основной дольковой архитектуры печени. При неонатальном гепатите обнаруживаются признаки тяжелого диффузного гепатоцеллюлярного заболевания: нарушение архитектуры долек, выраженная воспалительная инфильтрация и очаговый гепатоцеллюлярный некроз.
Желчные протоки при этом изменяются незначительно. Гигантоклеточная трансформация может обнаруживаться у младенцев с любым из этих заболеваний: признак не считается специфичным.
Гистологические изменения, характерные для пациентов с идиопатическим неонатальным гепатитом, могут встречаться и при др. заболеваниях: дефиците α1-антитрипсина, галактоземии, различных формах в/пе-ченочного холестаза. Снижение числа в/печеночных желчных протоков может выявляться при биопсии печени уже в первые несколько недель жизни. Но более характерную картину у таких пациентов можно обнаружить, если выполнить биопсию несколько позднее.
- Ведение пациентов с подозрением на билиарную атрезию. Всем пациентам с подозрением на билиарную атрезию должна быть выполнена диагностическая лапаротомия и прямая холангиография для поиска обструкции и определения ее локализации. Пациентам с потенциально корректируемыми поражениями м.б. выполнено прямое дренирование. Если же корректируемое поражение обнаружить не удалось, необходимо исследовать замороженные срезы тканей, полученных из области ворот печени. В ходе этого исследования можно обнаружить желчный эпителий, определить размеры и оценить проходимость остаточных желчных протоков. В некоторых случаях по результатам холангиограммы можно сделать вывод о том, что желчные протоки проходимы, их калибр уменьшен. В такой ситуации можно предположить, что холестаз обусловлен не облитерацией желчных путей, а малым их числом либо заметным снижением оттока желчи, связанным с в/печеночным заболеванием. В этих случаях следует избегать транссекции или дальнейшей диссекции в области ворот печени.
Пациентам, у которых не обнаружилось поддающегося коррекции поражения, следует выполнить операцию гепатопортоэнтеростомии (по Касаи). Ее необходимо провести, потому что в фиброзной ткани ворот печени могут присутствовать мельчайшие остатки желчных протоков — резидуальные каналы. Эти каналы м.б. напрямую связаны с системой в/печеночных желчных протоков. В таких случаях транссекция ворот печени с последующим наложением анастомоза между кишкой и проксимальной поверхностью транссекции позволяет дренировать желчь. Если отток желчи в срочном порядке не восстановить в течение первого месяца жизни, то в дальнейшем облитерация будет прогрессировать, и разовьется цирроз печени. Послеоперационное восстановление оттока желчи возможно, когда удается обнаружить проходимые микроскопические каналы с диаметром >150 мкм.
Также шанс успешного восстановления оттока желчи по результатам операции Касаи намного выше (90%), если она выполняется в возрасте до 8 нед. Необходимо своевременно обследовать младенцев с подозрением на билиарную атрезию и направлять их на лечение. Чтобы избежать запоздалой диагностики и добиться лучших клинических результатов, необходимо осуществлять обучение родителей, повышать осведомленность о заболевании среди мед. работников, шире внедрять карты для оценки окраски стула.
Операция Касаи в долгосрочной перспективе может принести пользу некоторым пациентам с некорригируемой билиарной атрезией. В большинстве случаев у них сохранится нарушение функции печени той или иной степени. У пациентов с билиарной атрезией часто наблюдается персистирующее воспаление в/печеночного билиарного дерева. Это позволяет предположить, что билиарная атрезия — отражение динамического процесса, вовлекающего всю гепатобилиарную систему. Этим можно объяснить развитие множества характерных для нее осложнений, напр. портальной гипертензии. Кратковременный «+» эффект гепатопортоэнтеростомии — декомпрессия и дренирование, достаточные для предотвращения развития цирроза и обеспечения процессов роста вплоть до момента успешной трансплантации печени. Применение ГКС после операции Касаи не сопровождалось увеличением показателей выживаемости пациентов или сохранности печени. Также не существует убедительных данных, позволяющих рекомендовать использование АБ или холеретических ЛП после операции.
7. Ведение хронического холестаза. При любой форме неонатального холестаза, вне зависимости от того, выступает ли в качестве первичного заболевания идиопатический неонатальный гепатит, в/печеночный холестаз или билиарная атрезия, у больных наблюдается повышенный риск прогрессирования хронического холестаза и развития его осложнений. Все вышеуказанное отражает степень остаточной функциональной способности печени и напрямую или косвенно ассоциируется со сниженным оттоком желчи. Любые в-ва, в норме выделяемые с желчью, задерживаются в печени и в последующем накапливаются в сыворотке крови и различных тканях. К таким в-вам относятся желчные кислоты, билирубин, холестерин и микроэлементы. Снижение поступления желчных кислот в проксимальный отдел кишечника приводит к нарушению переваривания и всасывания алиментарных длинноцепочечных триглицеридов и жирорастворимых витаминов.
Нарушение метаболической функции печени может изменить гормональный баланс и повлиять на утилизацию питательных в-в. Прогрессирующее повреждение печени может привести к развитию билиарного цирроза, портальной гипертензии и печеночной недостаточности.
Лечение пациентов с холестазом — эмпирическое. Им требуется тщательное наблюдение (табл. 7). Не существует эффективных методов терапии, способных остановить прогрессирование холестаза или предотвратить дальнейшее гепатоцеллюлярное повреждение и развитие цирроза. Серьезная проблема — нарушение роста, частично связанное с мальабсорбцией и мальнутрицией. Эти патологии — последствие нарушения переваривания и всасывания алиментарных жиров. Использование формул, содержащих триглицериды со средней длиной цепи, помогает улучшить баланс калорий. У детей с гепатобилиарными заболеваниями и хроническим холестазом в более позднем возрасте (при условии их выживания) может развиться дефицит жирорастворимых витаминов (A, D, Е и К). Напр., часто встречаются метаболические заболевания костей. Важно контролировать уровень жирорастворимых витаминов у таких пациентов.
Дегенеративный нервно-мышечный синдром обнаруживается у пациентов с хроническим холестазом, обусловленным дефицитом витамина Е. Для таких детей характерна прогрессирующая арефлексия, мозжечковая атаксия, офтальмоплегия и снижение вибрационной чувствительности. Специфические морфологические изменения обнаруживаются в ЦНС, периферических нервах и мышцах. Эти поражения можно предотвратить, поэтому они наблюдаются редко. Даже при условии развития эти поражения потенциально обратимы у детей <3-4 лет. Для этой группы пациентов характерна низкая концентрация витамина Е в сыворотке, повышенный перекисный гемолиз и низкий показатель отношения сывороточного витамина Е к общему содержанию липидов в сыворотке (<0,6 мг/г для детей <12 лет и <0,8 мг/г для детей более старшего возраста). Дефицит витамина Е можно предотвратить путем его перорального приема в больших дозах (до 1000 МЕ/сут). Пациентам, не способным усвоить достаточное количество витамина Е, может потребоваться пероральное введение сукцината D-α-токоферилполиэтиленгликоля 1000.
Для определения эффективности терапии необходимо контролировать уровень витамина Е в сыворотке.
Кожный зуд — особенно неприятное осложнение хронического холестаза, сопровождающееся появлением ксантом. Оба симптома связаны с накоплением холестерина и желчных кислот в сыворотке крови и тканях. Выведение этих соединений нарушается при закупорке желчных протоков. Но когда желчные протоки хоть сколько-нибудь проходимы, прием урсодезоксихолевой кислоты может способствовать увеличению оттока желчи или прерыванию энтерогепатической циркуляции желчных кислот и уменьшению количества ксантом и выраженности кожного зуда (см. табл. 7). Путем использования урсодезоксихолевой кислоты также можно добиться снижения уровня холестерина в сыворотке. Рекомендуемая начальная доза — 15 мг/кг в сутки. Также исследуется механизм ингибирования апикального натрий-зависимого переносчика желчных кислот. Использование этого механизма позволило бы предотвратить реабсорбцию желчных кислот в терминальном отделе подвздошной кишки, и могло бы оказаться эффективным в плане облегчения зуда и улучшения качества жизни больных.
Частичное внешнее билиарное отведение эффективно в лечении кожного зуда, рефрактерного к медикаментозной терапии, и позволяет добиваться хороших результатов в группе пациентов с хроническим холестазом без цирроза печени. Хирургическая техника заключается в резекции сегмента кишки, который в дальнейшем будет использован в качестве желчного канала. Один конец канала прикрепляют к желчному пузырю, а др. выводят на кожу, формируя стому. Главный недостаток методики — необходимость использовать стомный мешок. Открытая кнопочная холецистостомия и лапароскопическое частичное внешнее отведение желчных путей — модифицированные хирургические методики, которые также эффективны в плане облегчения зуда. Успешно применялась операция исключения подвздошной кишки. Она оказалась менее эффективной по сравнению с частичным внешним отведением желчных путей. Для пациентов с сохраняющимся зудом до расчесов кожи единственным вариантом остается трансплантация печени.
Прогрессирующий фиброз и цирроз печени приводят к развитию портальной гипертензии, асциту и кровотечению из варикозно расширенных вен. Наличие асцита — фактор риска развития спонтанного бактериального перитонита. Первый шаг в лечении пациентов с асцитом — исключить спонтанный бактериальный перитонит и ограничить потребления натрия до 0,5 г (~1-2 мг экв/кг в сутки). У пациентов с адекватным диурезом нет необходимости в ограничении жидкости. Если вышеуказанные меры неэффективны, показано назначение диуретиков. Предпочтительный диуретик — спиронолактон (1-3,3 мг/кг в сутки внутрь или Q12H). Если асцит не поддается контролю с помощью монотерапии спиронолактоном, следует добавить др. диуретик, напр. гидрохлортиазид или фуросемид. У пациентов с асцитом без периферических отеков на фоне терапии диуретиками наблюдается повышенный риск снижения объема циркулирующей плазмы и показателей диуреза.
Напряженный асцит влияет на почечный кровоток и системную гемодинамику. Парацентез и в/в-инфузии альбумина человека («Альбумина») могут улучшить гемодинамику, почечную перфузию и клиническую симптоматику. Дальнейшее наблюдение за пациентами включает диетическое консультирование и мониторинг уровней электролитов в сыворотке крови и моче.
У пациентов с портальной гипертензией часто наблюдаются кровотечения из варикозно расширенных вен и гиперспленизм. В этих случаях важно установить причину кровотечения, поскольку эпизоды ЖКК у пациентов с хроническими заболеваниями печени м.б. обусловлены гастритом или ЯБ. Лечебная тактика при этих состояниях отличается: поэтому перед началом терапии необходимо провести ДД с помощью эндоскопии. Если у пациента наблюдается снижение ОЦК, гемотрансфузию следует проводить с осторожностью, так избыточные объемы жидкости могут усугубить кровотечение. Детям не рекомендуется выполнять балонную тампонаду из-за риска развития тяжелых осложнений процедуры. Склеротерапия или эндоскопическое лигирование варикозно расширенных вен — потенциально эффективные в плане остановки кровотечения паллиативные вмешательства. Их выполнение — лучший вариант по сравнению с альтернативой в виде хирургических методик.
Показатели успешных трансплантаций печени у пациентов с запущенным заболеванием печени — >90%. Если операция технически осуществима, она продлит пациенту жизнь и поможет скорректировать метаболические нарушения при таких заболеваниях, как дефицит α1-антитрипсина, тирозинемия и болезнь Вильсона. В успехе операции большую роль играет адекватная тактика в интраоперационном, предоперационном и послеоперационном периодах, а также верное и осторожное применение иммунодепрессантов. Нехватка доноров печени малых размеров ограничивает применение трансплантации печени у младенцев и детей. Использование трансплантатов уменьшенных размеров трансплантации от живых доноров расширяет возможности успешного лечения детей младшего возраста.
8. Прогноз. Вариабельность прогноза у пациентов с идиопатическим неонатальным гепатитом отражает гетерогенность самого заболевания. В спорадических случаях 60-70% пациентов выздоравливают, и у них не наблюдается признаков структурных или функциональных нарушений печени. У 5-10% развивается стойкий фиброз или воспаление, у еще меньшего числа — тяжелое заболевание печени, напр. цирроз. Младенцы часто умирают еще на начальных этапах развития заболевания от кровотечения или сепсиса. Среди младенцев с семейным идиопатическим неонатальным гепатитом выздоравливают лишь 20-30%, у 10-15% развивается хроническое заболевание печени и цирроз. Им может потребоваться трансплантация печени.
б) Холестаз у детей старшего возраста. Холестаз, развивающийся после окончания неонатального периода, чаще всего обусловлен острым вирусным гепатитом или воздействием гепатотоксичных ЛП. Состояния, которые становятся причинами холестаза в неонатальном периоде, могут обусловить развитие хронического холестаза и у пациентов более старшего возраста. Поэтому детей старшего возраста и подростков с конъюгированной гипербилирубинемией необходимо обследовать на предмет острого и хронического вирусного гепатита, дефицита α1-антитрипсина, болезни Вильсона, заболеваний печени, ассоциированных с ВЗК, склерозирующего холангита, аутоиммунного гепатита, лекарственного поражения печени и синдромов в/печеночного холестаза. К прочим причинам относят обструкцию, обусловленную ЖКБ, опухолями БП, увеличением ЛУ или воспалением печени вследствие приема ЛП. Лечение холестаза у детей старшего возраста аналогично лечению холестаза у новорожденных (см. табл. 7).
Видео этиология, патогенез желтухи (повышения билирубина)