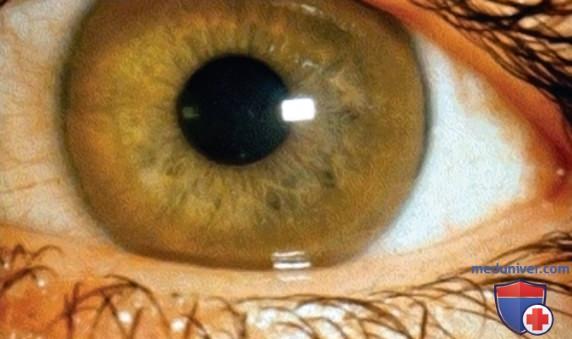
Болезнь Вильсона (гепатолентикулярная дегенерация) — это АуР-заболевание, которое м.б. связано с заболеваниями печени, дегенеративными изменениями в ГМ и кольцами Кайзера-Флейшера (Fleischer Bruno Otto; Kayser Bernhard) в роговице (рис. ниже). Заболеваемость составляет 1/30 000 рождений во всем мире. Существует специфическое лечение, однако это заболевание прогрессирует и может привести к летальному исходу, если его не лечить.
Кольцо Кайзера-Флейшера. По внешнему краю роговицы наблюдается коричневое окрашивание из-за отложения меди в десцеметовой оболочке. Здесь оно хорошо видно на фоне светло-зеленой радужки. Для безопасного обнаружения требуется осмотр при помощи щелевой лампы.
Оперативная диагностическая оценка болезни Вильсона у всех пациентов >5 лет, страдающих любой формой заболевания печени, облегчает оперативное начало лечения заболевания, соответствующее генетическое консультирование, скрининг родственников первой степени, а также позволяет проводить соответствующее лечение невильсоновской болезни печени после исключения интоксикации медью.
а) Патогенез. Аномальный ген болезни Вильсона обнаружен на хромосоме 13 (13q14.3) и кодирует β-полипептид медь-переносящей АТФазы) (АТР7В), транспортирующий медь аденозинтрифосфатазу P-типа (АТФазу), которая в основном экспрессируется в гепатоцитах и имеет решающее значение для выведения меди из желчных путей и включения меди в церулоплазмин.
Отсутствие или нарушение работы АТР7В приводит к снижению экскреции меди с желчью и диффузному накоплению меди в цитозоле гепатоцитов. Со временем клетки печени перегружаются, и медь перераспределяется в др. ткани, включая ГМ и почки, вызывая токсичность, в первую очередь в качестве мощного ингибитора ферментативных процессов.
Ионная медь ингибирует пируватоксидазу в ГМ и АТФазу в мембранах, что приводит к снижению содержания АТФ-фосфокреатинина и калия в тканях.
Было выявлено >500 мутаций, из которых >380 имеют подтвержденную роль в патогенезе заболевания; при генетическом тестировании можно идентифицировать патологический вариант. Большинство пациентов являются сложными гетерозиготами. Мутации, которые отменяют функцию гена, связаны с появлением симптомов заболевания уже в возрасте 3 лет, когда болезнь Вильсона обычно не учитывается при ДД.
Более мягкие мутации м.б. связаны с неврологическими симптомами или заболеваниями печени в возрасте 80 лет. Наиболее часто встречающиеся патогенные мутации АТР7В приводят к образованию белка, который связывает медь, но не может эффективно транспортироваться к апикальной поверхности гепатоцитов для выполнения своей функции экспорта меди. Фармакологическое ингибирование сигнальных путей р38 и Jun N-концевой киназы, активированной митогеном протеинкиназы in vitro, может избавить от этого дефекта и стать потенциальной новой терапевтической мишенью.
б) Клинические проявления. Формы поражения печени при болезни Вильсона включают бессимптомную гепатомегалию (со спленомегалией или без нее), подострый или хронический гепатит и острую печеночную недостаточность (с гемолитической анемией или без нее). Криптогенный цирроз печени, портальная гипертензия, асцит, отеки, кровотечение из варикозно расширенных вен или др. последствия печеночной дисфункции (задержка полового созревания, аменорея, нарушения свертываемости крови) м.б. проявлениями болезни Вильсона.
Проявления болезни изменчивы, с тенденцией к в/семейной модели. Заболевание печени является наиболее распространенным проявлением заболевания у детей и может предшествовать неврологическим симптомам <10 лет. Женщины в 3 раза чаще, чем мужчины, страдают острой печеночной недостаточностью. Когда болезнь Вильсона проявляется >20 лет, неврологические симптомы являются наиболее распространенным проявлением.
Неврологические расстройства могут развиваться незаметно или стремительно, с интенционным тремором, дизартрией, ригидной дистонией, паркинсонизмом, хореиформными движениями, отсутствием координации движений, ухудшением успеваемости в школе, психозом или изменениями в поведении. Кольца Кайзера-Флейшера отсутствуют у молодых пациентов с поражением печени при болезни Вильсона <50% случаев, но присутствуют у 95% пациентов с неврологическими симптомами.
Психические проявления включают депрессию, изменения личности, тревогу, обсессивно-компульсивное поведение или психоз.
Кумбс (Coombes Robert) - отрицательная гемолитическая анемия м.б. начальным проявлением, возможно, связанным с высвобождением большого количества меди из поврежденных гепатоцитов; эта форма болезни Вильсона без трансплантации обычно приводит к летальному исходу. Во время гемолитических эпизодов экскреция меди с мочой и уровень свободной меди в сыворотке крови заметно повышаются.
Могут присутствовать проявления почечного синдрома Фанкони (Fanconi Guido) и прогрессирующей ОПН с изменениями в канальцевом транспорте аминокислот, глюкозы и мочевой кислоты. Нетипичные проявления включают артрит, панкреатит, нефролитиаз, бесплодие или привычное невынашивание, ГКМП и гипопаратиреоз.
в) Патология. Все степени повреждения печени встречаются у пациентов с болезнью Вильсона, при этом наиболее часто встречаются стеатоз, баллонная дистрофия гепатоцитов, гранулы гликогена, минимальное воспаление и увеличенные клетки Купфера (Kupffer Karl Wilhelm von). Самой ранней гистологической особенностью болезни Вильсона является умеренный стеатоз, который может имитировать НАЖБП или неалкогольный стеатогепатит. Кроме того, поражение м.б. неотличимо от поражения аутоиммунным гепатитом.
При прогрессирующем повреждении паренхимы развиваются фиброз и цирроз печени. Ультраструктурные изменения в первую очередь затрагивают митохондрии и включают увеличение плотности матриксного материала, включения липидов и гранулированного материала, а также увеличение в/кристального пространства с расширением кончиков крист.
г) Диагноз. Болезнь Вильсона следует рассматривать у детей и подростков с необъяснимыми острыми или хроническими заболеваниями печени, неврологическими симптомами неизвестной причины, острым гемолизом, психическими заболеваниями, изменениями поведения, синдромом Фанкони (Fanconi Guido) или необъяснимыми заболеваниями костей (остеопороз, переломы) или мышц (миопатия, артралгия). Клиническое подозрение подтверждается исследованием показателей обмена меди.
У большинства пациентов с болезнью Вильсона уровень церулоплазмина в сыворотке крови снижен (<20 мг/дл). Неспособность меди быть включенной в церулоплазмин приводит к белку плазмы с более коротким периодом полураспада и, следовательно, к снижению стационарной концентрации церулоплазмина в циркуляции. Уровни церулоплазмина в сыворотке крови следует интерпретировать с осторожностью. Острые воспалительные состояния и повышенный уровень эстрогена (беременность, гормональная терапия или использование КОК) могут ложно повышать уровень церулоплазмина.
Кроме того, сывороточный церулоплазмин м.б. низким при аутоиммунном гепатите, целиакии, семейной ацерулоплазминемии или у носителей мутаций АТР7В легкие варианты болезни Менкеса (Menkes Ghosh M): синдром затылочного рога), у которых не проявляется болезнь перегрузки медью. Уровень свободной меди в сыворотке крови м.б. повышен на ранних стадиях болезни Вильсона (>1,6 мкмоль/л), а экскреция меди с мочой (обычно <40 мкг/сут) увеличивается до >100 мкг/сут и часто до 1000 мкг или более в день. Типичная экскреция меди с мочой у пациентов с нелеченной болезнью Вильсона составляет >1,6 мкмоль/сут у взрослых и >0,64 мкмоль/сут у детей.
В сомнительных случаях реакция выделения меди из мочи на хелатирование может помочь при диагностике. Перед суточным сбором мочи пациентам дают d-пеницилламин внутрь 250 мг 2 р/сут* Q12H; пациенты выделяют >1600 мкг/сут.
P.S. * Федеральные Клинические рекомендации по диагностике и лечению болезни Вильсона-Коновалова (гепатолентикулярная дегенерация), 2015 г.
Выявление колец Кайзера-Флейшера, которые могут отсутствовать у детей младшего возраста, требует осмотра офтальмологом с помощью щелевой лампы. После адекватного лечения кольца Кайзера-Флейшера рассасываются. При биопсии печени можно определить степень и тяжесть заболевания печени, а также измерить содержание меди в печени (обычно <10 мкг/г), но биопсия требуется только в том случае, если клинические признаки и неинвазивные тесты не позволяют поставить окончательный диагноз или если подозревается др. заболевание печени.
Накопление меди в печени является отличительной чертой болезни Вильсона, и измерение концентрации меди в паренхиме печени является методом выбора для диагностики. Содержание меди в печени >250 мкг/г (>4 мкмоль/г) является лучшим биохимическим доказательством болезни Вильсона, но снижение порога до 1,2 мкмоль/г улучшает чувствительность без существенного влияния на специфичность. Промежуточные уровни меди в печени могут присутствовать у бессимптомных носителей.
На более поздних стадиях болезни Вильсона содержание меди в печени м.б. сомнительным, поскольку цирроз печени приводит к неоднородному распределению меди в печени и ошибке отбора проб.
Родственники пациентов с болезнью Вильсона 1-й степени должны быть обследованы на наличие бессимптомного (предсимптомного) заболевания. Этот скрининг должен включать определение уровня церулоплазмина в сыворотке крови и суточную экскрецию меди с мочой. Если эти результаты являются патологическими или сомнительными, следует провести биопсию печени для определения морфологии и содержания меди в печени.
Генетический скрининг с помощью анализа связей или прямого анализа мутаций ДНК возможен, особенно если мутация для случая пробанда известна или пациент из области, где распространена специфическая мутация, напр. в Центральной и Восточной Европе, где мутация H1069Q присутствует у 50-80% пациентов.
д) Лечение. После постановки диагноза болезни Вильсона следует начать пожизненное лечение, направленное на ограничение поглощения меди и стимулирование выведения меди с помощью диетических и фармакологических мер. Нормальная диета содержит 2-5 мг меди в день. Для пациентов с болезнью Вильсона потребление меди с пищей должно быть ограничено до <1 мг/сут. Следует избегать продуктов с высоким содержанием меди, таких как печень, моллюски, орехи и шоколад. Если содержание меди в питьевой воде превышает 0,1 мг/л, может потребоваться деминерализация воды.
Начальным лечением у симптоматических пациентов является введение хелатирующих медь агентов, что приводит к быстрому выведению избытка накопившейся меди. Хелатная терапия проводится следующими ЛП: D-пеницилламин (β,β-диметилцистеин) внутрь 1 г/сут в 2 приема перед едой для взрослых и 20 мг/кг в сутки для педиатрических пациентов или дигидрохлорид триэтилентетрамин (триен, ТЕТА, триентин) внутрь 0,5-2,0 г/сут для взрослых и 20 мг/кг в сутки для детей. В ответ на хелатирование экскреция меди с мочой увеличивается с заметным улучшением печеночной и неврологической функции и исчезновением колец Кайзера-Флейшера.
У 10-50% пациентов, первоначально получавших пеницилламин по поводу неврологических симптомов, наблюдается ухудшение их состояния. Токсические эффекты пеницилламина проявляются в 10-20% случаев и состоят из реакций гиперчувствительности (напр., синдром Гудпасчера (Goodpasture Ernest William), СКВ и полимиозит), взаимодействия с коллагеном и эластаном, дефицита др. элементов, таких как цинк, а также апластической анемии и нефроза. Поскольку пеницилламин является антиметаболитом витамина В6, необходимы дополнительные количества этого витамина. По этим причинам триентин является предпочтительной альтернативой и считается терапией первой линии для некоторых пациентов.
Триентин имеет мало известных побочных эффектов. Тетратиомолибдат аммония является еще одним альтернативным хелатирующим агентом, изучаемым у пациентов с неврологическим заболеванием; первоначальные результаты показывают, что значительно меньше пациентов испытывают неврологическое ухудшение при применении этого ЛП по сравнению с пеницилламином. Начальная доза составляет 120 мг/сут (20 мг между приемами пищи 3 р/сут и 20 мг во время еды 3 р/сут). Побочные эффекты включают анемию, лейкопению, тромбоцитопению и умеренное Т АЛТ и ACT. Тетратиомолибдат аммония также обладает антиангиогенным действием благодаря своему обширному обезболивающему эффекту.
Цинк также использовался в качестве адъювантной терапии, поддерживающей терапии или первичной терапии у пациентов с предсимптомной формой заболевания из-за его уникальной способности ухудшать всасывание меди в ЖКТ. Цинка ацетат можно давать взрослым в дозе 25-50 мг элементарного цинка 3 р/сут и 25 мг 3 р/сут детям >5 лет. Побочные эффекты в основном ограничиваются раздражением желудка, но также включают снижение хемотаксиса лейкоцитов и повышение уровня липазы и/или амилазы в сыворотке крови.
В современных КР рекомендуют, чтобы все пациенты с симптомами болезни Вильсона получали хелатирующий агент (пеницилламин или триентин). Пациентам следует посоветовать не прекращать прием этих ЛП внезапно, т.к. внезапное прекращение терапии может привести к быстрому развитию болезни Вильсона. Цинк может играть роль в качестве терапии первой линии у пациентов с неврологическими заболеваниями, но исключительная монотерапия цинком при симптоматических заболеваниях печени спорна и не рекомендуется. Антиоксиданты (витамин Е и куркумин) и фармакологические шапероны (4-фенилбутират и куркумин) могут играть роль дополнительного лечения, но необходимы дополнительные исследования.
е) Прогноз. Нелеченые пациенты с болезнью Вильсона могут умереть от печеночных, неврологических, почечных или гематологических осложнений. Медикаментозная терапия редко эффективна у пациентов с острой печеночной недостаточностью. Прогноз для пациентов, которые быстро и постоянно получают пеницилламин, является переменным и зависит от времени начала и индивидуальной реакции на хелатирование. Трансплантация печени должна быть рассмотрена для пациентов с острой печеночной недостаточностью или декомпенсированным циррозом печени из-за болезни Вильсона.
Трансплантация печени при прогрессирующем неврологическом заболевании остается спорной. Трансплантация печени является лечебной, с 5-летней выживаемостью 85-90%. У бессимптомных братьев и сестер пострадавших пациентов раннее введение хелатной или цинковой терапии может предотвратить проявление заболевания.