Стержневые миопатии являются наиболее распространенной формой врожденной миопатии, состоящей из болезни центрального стержня, мультистержневой миопатии и атипичных стержней. Стержни — это области внутри мышечных волокон, в которых обнаружена только аморфная зернистая цитоплазма, без миофибрилл и органелл. Стержни лишены митохондрий, которые содержат неупорядоченные саркомерные белки.
Гистохимические пятна показывают отсутствие ферментативной активности всех типов в этих стержнях, а также отсутствие сократительных белков (актина и миозина), которые образуют тонкие и толстые миофиламенты. Продольно обширные области в центральной области миофибры, лишенные окислительной ферментативной активности, представляют собой центральные стержни, а множественные меньшие области пониженной активности, затрагивающие более короткие сегменты миофибрилл, характерны для мульти- и мини-стержней.
При электронной микроскопии стержни характеризуются аномальной саркомерной структурой, включающей поток Z-диапазона, полную миофибриллярную дезорганизацию и накопление материала Z-диапазона. Хотя варианты центральных стержней, называемые мульти- и мини-стержнями, описаны в некоторых семействах, считается, что они представляют один и тот же основной патологический процесс. Патологические особенности могут эволюционировать, причем изменения и аномалии становятся все более очевидными с течением времени.
а) Клинические проявления. Фенотипический спектр основных миопатий колеблется от легкой до тяжелой. Гипотония, слабость суставов, задержка двигательного развития, слабость ТБС или аксиальных мышц, ортопедические осложнения, такие как рецидивирующие вывихи плеча или надколенника, врожденный вывих бедра либо дисплазия или деформации стопы, могут представлять собой признаки. У детей старшего возраста болезнь центрального стержня является важным ДД прогрессирующего грудопоясничного сколиоза. Существует также в/семейная вариабельность, причем у некоторых людей наблюдаются только мышечная ригидность, миалгия при ФН или рабдомиолиз.
Генетическое разрешение основных миопатий привело к мутационно-специфическим клиническим проявлениям и корреляциям фенотип-генотип.
1. Болезнь центрального стержня. Болезнь центрального стержня чаще всего ассоциируется с мутациями гена рианодина 1 (RYR1), которые описываются как преобладающие генетические причины недистрофических нервно-мышечных расстройств. Эти расстройства варьируют от доминантно унаследованных болезни центрального стержня, подгрупп рецессивно наследуемых мультиминикорных заболеваний (мультистержневая миопатия), центронуклеарной миопатии и врожденной диспропорции мышечных волокон до признака восприимчивости к злокачественной гипертермии.
Злокачественная гипертермия — это доминантно наследуемый аллельный признак, описываемый как фармакогенетическая предрасположенность к тяжелой и потенциально опасной для жизни реакции в ответ на галогенированные анестетики и деполяризующие миорелаксанты. Злокачественную гипертермию можно подозревать у человека с врожденной миопатией, когда (1) имеется положительный семейный анамнез злокачественной гипертермии, (2) ранее были трудности с анестезией и (3) у пациента есть документированная мутация RYR1.
Доминантно наследуемая болезнь центрального стержня, связанная с RYR1, характеризуется легкой и умеренной мышечной слабостью, проявляющейся от младенчества до детства (рис. 1). Клинический спектр колеблется от последовательности деформации акинезии плода до более мягких форм у взрослых. Распределение мышечной слабости обычно проксимальное, с заметным вовлечением ТБС и аксиальных мышц. Врожденный вывих бедра, сколиоз и генерализованная слабость суставов встречаются часто.
Рисунок 1. Заболевание центрального стрежня. Фотография близнецов, один из которых болен. Обратите внимание на слабость проксимальных отделов верхних конечностей.
В отличие от рецессивных форм с более выраженным клиническим фенотипом, вовлечение экстраокулярных мышц отсутствует. Бульбарное, респираторное и сердечное поражение встречается редко. Миалгия м.б. заметной. За исключением пациентов с тяжелым неонатальным началом, большинство пациентов с болезнью центрального стержня самостоятельно передвигаются. Болезнь центрального стержня стабильна в течение длительного периода времени, с возможным медленно прогрессирующим течением во взрослом возрасте. RYR1-родственная злокачественная гипертермия аллельна болезни центрального стержня, и некоторые пациенты с болезнью центрального стержня также м.б. восприимчивы к злокачественной гипертермии.
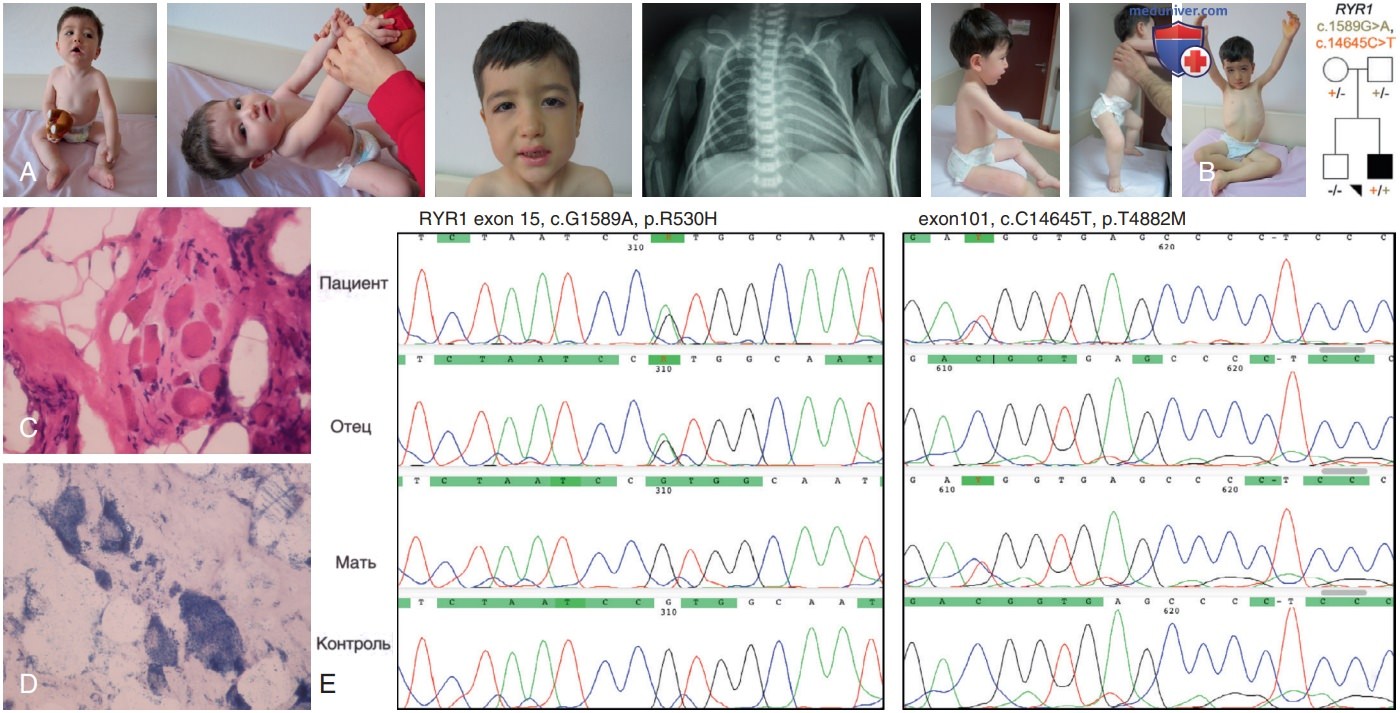
На рис. 2 показана семья с рецессивной мутацией RYR1 у индексного пациента и его бессимптомный отец, несущий доминантный ген RYR1 -злокачественной гипертермии. Характеристики и недавно выявленные фенотипы, обусловленные мутациями RYR1, суммированы в табл. 6.
Рисунок 2. Индекс пациента с задержкой развития и гипотонией в возрасте 19 мес. Обратите внимание на двусторонние переломы плечевой кости при рождении. У него был предыдущий диагноз несовершенного остеогенеза. У него слабость лица, миопатическое лицо, вовлечение мышц-сгибателей шеи с отставанием головы; его максимальная двигательная способность — сидеть без опоры: A — он не может стоять на ногах; B — в возрасте 4 лет пациент не может ходить; C — биопсия мышц в возрасте 19 мес, демонстрирующая миопатические изменения с повышенной инфильтрацией жировой и фиброзной ткани (НЕ); D — центральных ядер (NADH). Индексный пациент несет рецессивную мутацию RYR1, а его отец — доминантную мутацию RyR1-предрасположенность к злокачественной гипертермии (E)
Мутации RYR1, связанные с злокачественной гипертермией, также были описаны как распространенная причина индуцированных и эпизодических фенотипов, таких как рабдомиолиз при ФН, на долю которых приходится до 30% проявлений у ЗЛ в течение всей жизни. Поздние проявления в зрелом возрасте подчеркивают актуальность врожденных миопатий для взрослых. Предрасполагающий генетический фон следует учитывать, если эпизоды являются семейными, рецидивирующими, вне контекста выполняемого упражнения или предшествуют др. симптомам, таким как судороги, миалгия и слабость. Рабдомиолиз, связанный с RYR1, может возникать до 72 ч после ФН и имитировать вирусный миозит; в отличие от др. метаболических миопатий, голодание, по-видимому, не является пусковым фактором.
Благодаря экспрессии рецепторов рианодина, отличных от поперечнополосатой скелетной мышцы, распознаются нескелетные мышечные проявления миопатий, связанных с RYR1. Легкие нарушения кровоточивости описаны у пациентов со злокачественной гипертермией, несущих мутации усиления функции RYR1 путем изменения функции гладкомышечных клеток сосудов. Дефект кровотечения в животной модели и у одного пациента был отменен лечением антагонистом RyR1 дантроленом, что предполагает терапевтическую роль для связанных с RYR1 нарушений кровотечения и, возможно, др. Др. наблюдаемым фенотипом является тяжелое поражение ЦНС у подростка, страдающего эпизодом злокачественной гипертермии. Поразительное сходство в плане поражения мозжечка, наблюдаемое у этого пациента и жертв теплового инсульта, указывало на потенциальную связь между RyR1-связанным рабдомиолизом и нейролептическим злокачественным синдромом.
Некоторые психофармакологические ЛП, такие как оланзапин, следует рассматривать в качестве триггерных агентов у пациентов с мутациями RYR1 и рабдомиолизом при ФН. Новым вопросом является вовлечение сердца в миопатии, связанные с RYR1. Описаны внезапная необъяснимая смерть, ДКМП, предположительно вызванная вирусной инфекцией, двустворчатый аортальный клапан и синусовая брадикардия; кардиологическая оценка должна рассматриваться для определения сердечного фенотипа с миопатиями, связанными с RYR1.
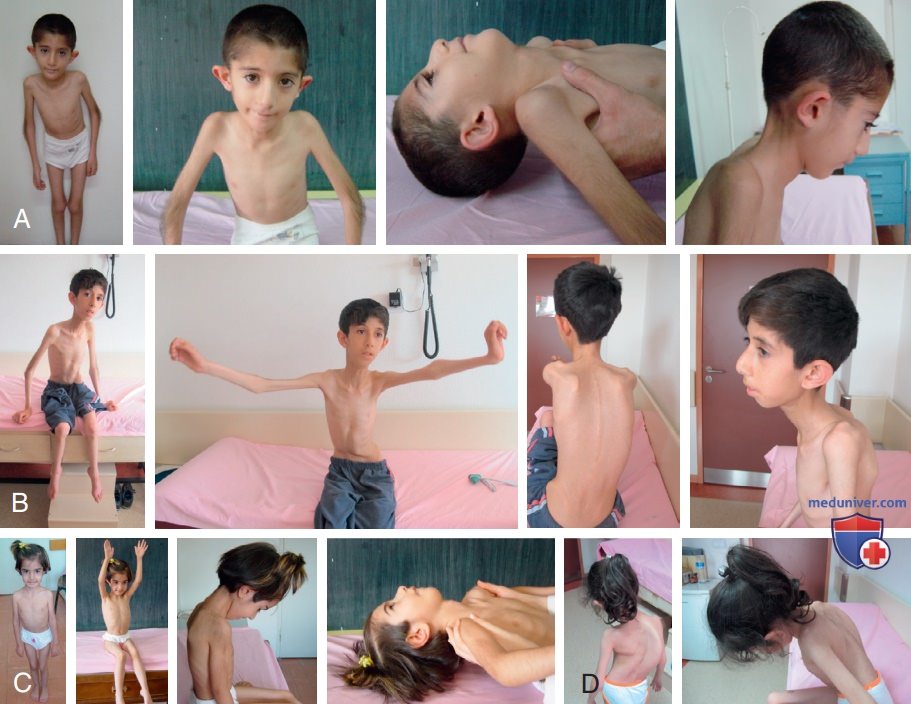
2. Мульти/мини-стержневая миопатия. Миопатия с мульти/мини-стержнями обычно наследуется рецессивно, и клинический фенотип зависит от лежащего в основе генетического фона. Симптоматика может варьировать и перекрываться между наиболее распространенной и узнаваемой классической формой, тяжелой неонатальной формой, формой с внешней офтальмоплегией и умеренной формой с вовлечением рук. Классический фенотип, обусловленный рецессивными мутациями селенопротеина N1 (SEPN1), можно суммировать как осевую слабость, раннюю ригидность позвоночника, сколиоз и ДН (рис. 3). Начало заболевания раннее, с преимущественным вовлечением мышц шеи. Младенцы, не способные держать голову, несмотря на способность самостоятельно ходить, имеют изолированную миопатию шеи или синдром опущенной головы.
Рисунок 3. Пациенты с типичным фенотипом мультимини-стержневой миопатии, связанной с SEPN1 (MmD); A — в возрасте 10 лет; B — 12 лет; C — 7 лет; D — 8 лет. Обратите внимание на астенический, атрофический фенотип, с ригидным синдромом позвоночника, слабостью мышц-сгибателей шеи и различной степенью сколиоза
Миопатическое лицо, высоко изогнутое или «волчье» нёбо, высокий голос, трудности с питанием и неспособность развиваться м.б. сопутствующими чертами. Эти пациенты м.б. очень похожи друг на друга, с астеническим и атрофическим мышечным фенотипом и задержкой роста, которые в основном исследуются и направляются с предварительным диагнозом целиакии. Сильнее поражаются проксимальные мышцы плечевого пояса и внутренняя поверхность бедра. Осевая слабость со временем сменяется контрактурами мышц-разгибателей позвоночника, что приводит к ригидной деформации позвоночника. Обычно ко второй декаде наблюдаются прогрессирующий сколиоз, боковое отклонение туловища и нарушение дыхания, что в целом несоразмерно слабости скелетных мышц. Мульти/мини-стержневую миопатию следует учитывать при ДД заболеваний, сопровождающихся ранней нервно-мышечной хронической ДН (т.е. хронической слабостью дыхательных мышц, когда пациент еще находится на амбулаторном этапе). Поражение ДП может привести к вторичной СН.
Офтальмоплегия не является особенностью этой классической формы, но она исключительно хорошо распознается на последних стадиях заболевания у пациентов с тяжелым течением.
Фенотипы мульти/мини-стержневой миопатии, обусловленные рецессивными мутациями RYR1, характеризуются более мягкими дыхательными, но выраженными бульбарными нарушениями по сравнению с таковыми при классической форме. Внешняя офтальмоплегия, рецидивирующие эпизоды периодического паралича, дистальная слабость и истощение, в основном поражающие руки, артрогрипоз, крипторхизм и дисморфические особенности также были описаны в спектре мульти/мини-стержневой миопатии, связанном с RYR1. ГКМП, связанная с дефицитом короткоцепочечной ацил-кофермента А дегидрогеназы, и первичные кардиомиопатии, обусловленные мутациями в генах тяжелой цепи миозина 7 (MYH7) или титина (TTN), были описаны в мульти/мини-стержневой миопатии.
Хотя о мульти/мини-стержневой миопатии, связанной с SEPN1, не сообщается, существует потенциальный риск развития МХС при этой миопатии, связанной с RYR1.
б) Лабораторные результаты. Диагностика стержневых миопатий м.б. сложной задачей и требует сочетания детальной клинической (распознавание фенотипа) и лабораторной (гистопатологическая, мышечная визуализация, генетическая) оценки и интерпретации. Значение КФК в сыворотке крови нормальное, за исключением кризов злокачественной гипертермии, которые могут привести к рабдомиолизу или обширному острому некрозу миофибры. Мышечная визуализация (УЗИ и МРТ) может служить неинвазивным инструментом для описания характерного избирательного поражения мышц. Распознавание этих паттернов может помочь отличить типичные доминантно наследованные формы болезни центрального стержня и связанные с SEPN1 мульти/мини-стержневой миопатией от различных нервно-мышечных заболеваний.
Диагноз стержневой миопатии, основанный на патологических данных, м.б. простым; однако типичная картина может развиваться с течением времени, когда ранние биопсии мышц почти не показывают или показывают минимальные изменения. Формирование стержня — это неспецифическая находка, которая может наблюдаться в процессе денервации, тенотомии, метаболических состояниях или даже у здоровых пробандов после интенсивных упражнений. Изъеденные волокна, описанные при мышечной дистрофии, могут напоминать мини-стержни при мульти/мини-стержневой миопатии D. Наличие стержней без сопутствующей слабости, как сообщалось, у некоторых пациентов с восприимчивостью к злокачественной гипертермии недостаточно для постановки диагноза стержневой миопатии. Стержни и др. структурные аномалии, характерные для др. структурных миопатий, таких как немалиновая миопатия или болезнь центрального стержня, могут сосуществовать.
Мышечные дистрофии, обусловленные мутациями ламина А/С (LMNA), миопатии, связанные с коллагеном VI, метаболические миопатии (болезнь Помпе (Pompe, Joannes Cassianus)), миофибриллярные миопатии у пациентов с кардиомиопатией и врожденные миастенические синдромы могут имитировать стержневые миопатии на основе клинических и/или патологических особенностей и должны учитываться при ДД.
Из-за крайних клинических и патологических совпадений между ранними мышечными заболеваниями произошел сдвиг в традиционных диагностических путях. Принимая во внимание все эти вопросы, диагностика основных миопатий, как и др. врожденных миопатий, требует совместных усилий со стороны клинициста, патолога и молекулярного генетика.
в) Генетика. Миопатии центрального стержня передаются либо как АуД, либо как АуР-признак, либо как доминантные мутации de novo. Они вызваны одним и тем же аномальным геном в локусе 19q13.1. Этот ген программирует рианодиновый рецептор (RYR1), тетрамерный рецептор, который содержит кальциевый канал без напряжения; он преобладает в саркоплазматическом ретикулуме и особенно в месте соединения Т-канальца с цистернами саркоплазматического ретикулума. Он содержит канал, по которому кальций выделяется среди миофиламентов.
Мутации в гене RYR1 также являются причиной злокачественной гипертермии. При центронуклеарных миопатиях АуР мутации RYR1, как известно, являются частой причиной, и недавно был также описан пациент с доминантной de novo мутацией RYR1. Пациенты с врожденной миопатией, птозом, внешней офтальмоплегией и видными внутренними ядрами в дополнение к др. структурным находкам являются весьма вероятными кандидатами на мутации RYR1. Следует отметить, что рецессивное заболевание стрежня м.б. связано с тканеспецифическим глушением нормального аллеля эпигенетическим феноменом. Некоторые миссенс-мутации м.б. связаны с АуД злокачественной гипертермией, и лечение бессимптомного носителя аллеля восприимчивости злокачественной гипертермии должно проводиться соответствующим образом.
Мутации в гене медленного β-миозина (MYH7), АуР мутации титина (TTN) и рецессивные мутации гена сателлитных клеток (MEGF10) являются др. идентифицированными причинами сердечной миопатии. Последняя характеризуется ранней миопатией, арефлексией, РДС и дисфагией (EMARRD). Описан один пациент с тяжелой врожденной миопатией и офтальмоплегией, а также рецессивными вариантами в гене, кодирующем субъединицу альфа-1 дигидропиридинового рецептора (CACNA1S), ген, в котором доминантные мутации, как известно, связаны с гипокалиемическим периодическим параличом и злокачественной гипертермией. Необходимы функциональные исследования, чтобы связать мутации CACNA1S с врожденными миопатиями.
Мультистержневая миопатия вызывается в основном рецессивными мутациями в SEPN1 и RYR1. Селенопротеин N — это интегральный мембранный белок, локализованный в эндоплазматическом ретикулуме, который экспрессируется в нескольких тканях, включая скелетную мышцу, сердце, легкие и плаценту. Он также сильно выражен в диафрагме; это может объяснить обнаружение ранней рестриктивной ДН у пациентов. Мутации SEPN1 также вызывают врожденную диспропорцию мышечных волокон и ригидную мышечную дистрофию позвоночника.
г) Лечение и прогноз. Лечение стержневых миопатий носит симптоматический характер и, как правило, должно проводиться параллельно с общепринятыми стандартными рекомендациями по лечению врожденных миопатий. Ортопедические осложнения, реабилитация и проблемы с питанием должны решаться соответствующим образом. Сколиоз и др. деформации скелета требуют особого внимания, поскольку они могут быстро развиваться и прогрессировать по степени тяжести непропорционально слабости конечностей. По сравнению с др. врожденными миопатиями наблюдается большее число неудач лечения при врожденном вывихе бедра и дисплазии при болезни центрального стержня.
Болезнь центрального стержня последовательно ассоциируется со злокачественной гипертермией, которая может предшествовать диагностике болезни центрального стержня. Все пациенты и бессимптомные носители должны быть проконсультированы с точки зрения потенциально фатальных побочных реакций на летучие анестетики и миорелаксанты. Следует рассмотреть возможность предоперационной анестезиологической консультации у пациентов, которые, как известно, подвергаются общей анестезии. Ношение мед. браслета оповещения должно быть рекомендовано в случае любой чрезвычайной ситуации. Лечение злокачественной гипертермии требует применения дантролена и дополнительных мер поддерживающей терапии. Следует отметить, что профилактический дантролен не рекомендуется применять перед анестезией даже в тех случаях, когда была установлена злокачественная гипертермия.
Возможно коварное начало поражения дыхательных мышц, особенно у пациентов с мутациями мульти/ми-нистержневой миопатии и SEPN1. У пациентов м.б. симптомы после интеркуррентного заболевания, анестезии или даже седации во время процедуры биопсии мышц. Междисциплинарный уход требует учета данных от врачей-пульмонологов. Признаки и симптомы нарушения сна — нарушение дыхания, синдром ночной гиповентиляции должны быть подвергнуты сомнению. Для своевременного введения неинвазивной ИВЛ с «+» давлением необходимы тесты дыхательной функции в положении сидя и лежа, а также полисомнография. Пациентам с тяжелым ранним началом заболевания может потребоваться инвазивная ИВЛ. Сердечные осложнения при врожденной хлоридной диарее встречаются редко, но в большинстве случаев целесообразно проведение базовых ЭКГ и ЭхоКГ исследований. Вторичная дисфункция ПЖ и СН может осложнить ситуацию у пациентов с нарушением дыхания.
Сообщалось о субъективном улучшении результатов мышечной силы и функциональных тестов у пациентов с болезнью центрального стрежня, принимающих β-2 агонисты (сальбутамол, альбутерол). Современные и будущие терапевтические подходы включают (1) модификацию функции RYR1, (2) коррекцию ассоциированных окислительных нарушений, (3) использование фармакологических соединений, усиливающих сократительную способность мышц и/или нервно-мышечную передачу, и (4) коррекцию специфического дефекта гена. Ацетилцистеин («N-ацетилцистеин») как антиоксидант может служить потенциальным вариантом лечения миопатий, связанных с RYR1 и SEPN1, и в настоящее время проводятся первые клинические испытания на людях.