Миофибриллярные миопатии — это редкие наследственные или спорадические прогрессирующие нервно-мышечные расстройства, диагностируемые на основании отчетливых морфологических признаков. Существует широкий спектр клинической и генетической гетерогенности в рамках миофибриллярной миопатии, которые также подразделяются на подгруппы как белковые агрегатные миопатии. Описаны разл. фенотипические проявления, обусловленные поражением сердечной, скелетной и гладкой мускулатуры.
Основные гистопатологические данные можно определить как очаговую дезинтеграцию миофибрилл преимущественно на уровне Z-диска, накопление продуктов деградации миофибрилл и эктопическую экспрессию большого количества белков. Растворение миофибрилл начинается на Z-диске, и некоторые саркомеры миофибрилл имеют дезорганизацию или растворение миофибрилл, прилегающих к др. областям нормальных саркомеров внутри того же волокна.
Аномальные скопления белков, интенсивная конгофилия многих гиалиновых структур, интернализованные ядра, расщепление волокон, вакуоли, ядроподобные поражения, легкое или сильное увеличение эндомизиального коллагена и повышенная вариабельность размеров волокон от очень гипотрофических до гипертрофических волокон являются одними из общих признаков. Эти зоны связаны с потоком Z-дисков, и в агрегатах наблюдается экспрессия большого количества белков, включая дистрофин, саркогликаны, убиквитин, промежуточные нити десмина, αВ-кристаллин и несколько белков Z-диска, таких как миотилин и филамин-С. Митохондриальная дисфункция в виде аномального распределения митохондрий является частым явлением.
Хотя детальное иммуноцитохимическое и ультраструктурное исследование мышечной ткани биопсии необходимо для постановки диагноза и может дать ключ к пониманию основного измененного гена, окончательный диагноз подтипа миофибриллярной миопатии зависит от молекулярно-генетического тестирования. Сверхэкспрессия или повышенная регуляция нормальных белков, таких как десмин или ав-кристаллин в миофибрах, м.б. дополнительной особенностью во многих др. нервно-мышечных состояниях, поэтому миофибриллярную миопатию следует использовать, когда эти накопления обусловлены мутацией в соответствующем белке. Подтипы миофибриллярной миопатии классифицируются в соответствии с пораженным белком: десминопатия, αВ-кристаллинопатия или Bag3 миопатия.
а) Клинические проявления и подтипы миофибриллярных миопатий в зависимости от генетического фона. Большинство миофибриллярных миопатий не имеют симптомов в детском возрасте, но иногда у детей старшего возраста и подростков проявляются ранние симптомы неспецифической проксимальной и дистальной слабости. Миофибриллярные миопатии обычно присутствуют в проксимальных и дистальных мышцах. Дистальная слабость обычно более выражена, чем проксимальная.
Сенсорные симптомы, скованность мышц, боль и судороги м.б. дополнительными симптомами. У больных могут наблюдаться признаки периферической невропатии и клинической кардиомиопатии. АуР-формы имеют более раннее и тяжелое течение по сравнению с АуД-формами. Существует также большая межсемейная и в/семейная вариабельность в клинической выраженности заболевания. Степень нарушений и характер прогрессирования варьируют у разных больных.
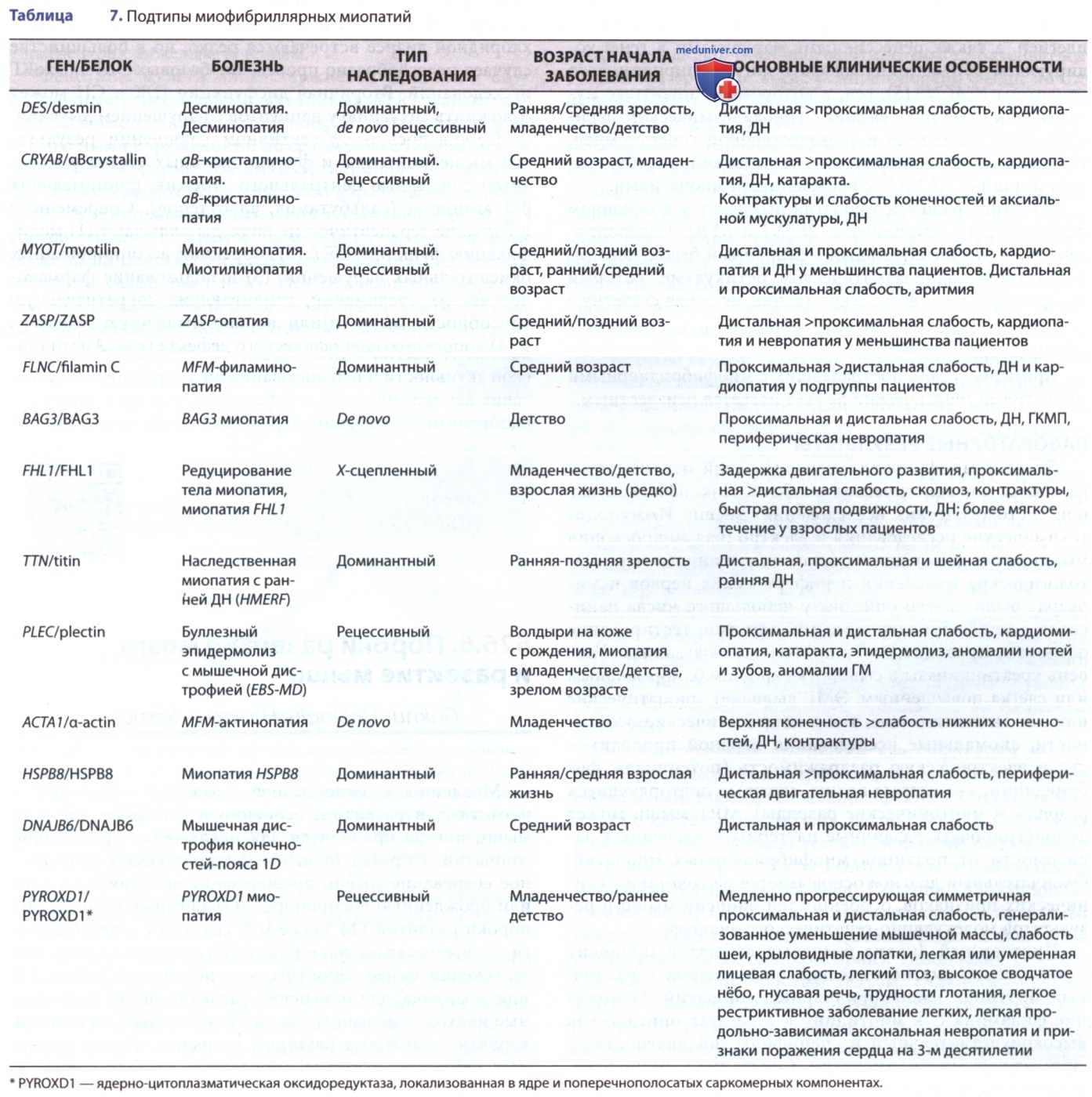
Подтипы миофибриллярных миопатий с основными клиническими признаками приведены в табл. 7. Поражение сердца иногда м.б. начальным и единственным симптомом, особенно при десминопатиях. Среди сердечных проявлений — синкопальные эпизоды, дефекты проводимости (полная АВ-блокада, БПНПГ, левый передний гемиблок), нарушения ритма (желудочковая аритмия), кардиомиопатия (ДКМП, РКМП, ГКМП), персистирующий ОАП и ЗСН. Вовлечение лицевых, осевых и шейных мышц, бульбарные расстройства, трудности с глотанием и кормлением, ригидная деформация позвоночника, ранняя ДН и ранняя катаракта м.б. дополнительными симптомами в диагностике. Поражение гладкой мускулатуры может проявляться в виде нарушения всасывания в кишечнике и псевдообструкции.
Некоторые подтипы миофибриллярных миопатий м.б. связаны с ранним инфантильным началом. Примером может служить уникальная АуР-миопатия у детей народности Кри*, характеризующаяся тяжелой генерализованной мышечной гипертонией, которая не купируется нервно-мышечной блокадой и, следовательно, имеет миопатическое происхождение.
P.S. * Кри (англ. Cree, фр. Cri, самоназвание — nehiyawak) — этническая общность в Северной Америке.
Большинство умирают в младенчестве от ДН из-за поражения диафрагмы. Биопсия мышц показывает сходные результаты со многими др. миофибриллярными миопатиями (рис. ниже); причиной является новая мутация гена αВ-кристаллина. Раннее начало заболевания также может наблюдаться при десминопатии; Bag3 миопатия; АуР буллезный эпидермолиз с мышечной дистрофией (EBS-MD) в группе плектинопатий; наследственная миопатия с ранней ДН (HMERF) в группе титинопатий; актин-родственная миофибриллярным миопатиям и PYROXD1 -миопатия.
Электронная микрофотография биопсии четырехглавой мышцы бедра девочки 1 мес с миофибриллярной миопатией Кри. Внутри того же миофибра некоторые саркомеры хорошо сформированы, а др. демонстрируют беспорядок толстых и тонких миофиламентов и фрагментацию Z-полос. Митохондрии выглядят нормальными (х21 400)
Гены болезни миофибриллярной миопатии кодируют белки, которые являются структурными и функциональными компонентами саркомера, экстрасаркомерного цитоскелета или систем контроля качества белка. PYROXD1 классифицируется как пиридиновая нуклеотид-дисульфидная оксидоредуктаза класса I, которая принадлежит к древнему семейству ферментов, регулирующих окислительно-восстановительное состояние др. белков. Описана ранняя PYROXD 1-миопатия, гистологически характеризующаяся множественными интернализованными ядрами, большими зонами саркомерной дезорганизации, скоплением тонких нитей, утолщенными Z-полосами и десмин «+» включениями.
Существует отличительная гистопатология, которая сочетает в себе черты центральной и мини-стержневой болезни и центронуклеарных, миофибриллярных и немалиновых миопатий у пациентов, описанных до сих пор, что ясно указывает на совпадение между врожденными миопатиями и миофибриллярными миопатиями.
Примерно у 50% пострадавших с миофибриллярными миопатиями генетический дефект остается неизвестным.
б) Лабораторные результаты. Диагноз миофибриллярных миопатий основывается на общих морфологических особенностях, наблюдаемых при гистологических исследованиях мышц. Иммуноцитохимические исследования и электронная микроскопия мышц могут дать ключ к разгадке причинного гена. Патологические изменения периферических нервов и миокарда были кратко описаны у небольшого числа пациентов с миофибриллярными миопатиями; тестирование обычно не проводится по клиническим показаниям. Уровень креатинкиназы в сыворотке крови м.б. нормальным или слегка повышенным.
ЭМГ выявляет миопатические или как миопатические, так и нейропатические особенности, аномальные исследования нервной проводимости и электрическую раздражимость (потенциалы фибрилляции, «+» острые волны, сложные повторяющиеся разряды и миотонические разряды). МРТ мышц может демонстрировать различные паттерны вовлечения в зависимости от подтипов миофибриллярных миопатий. Окончательный диагноз основывается на сочетании клинических признаков, особенностей биопсии мышц и результатов молекулярно-генетического анализа.
Протеомный анализ белковых агрегатов приводит к выявлению диагностических биомаркеров в различных подтипах миофибриллярных миопатий. Отношение филамина С к миотилину в агрегатах описано как высокочувствительный и специфический диагностический маркер миотилинопатии. Сочетание иммунофлуоресцентных исследований с протеомными данными еще больше облегчит идентификацию нескольких белков, участвующих в контроле качества и деградации белка, которые также могут выступать в качестве терапевтических мишеней.
ДД включает врожденные миопатии, миотоническую дистрофию, митохондриальные заболевания и периферические невропатии в детском возрасте.
в) Лечение. Патогенетической терапии нет. Лечение носит поддерживающий и симптоматический характер. Скрининг сердца (ЭКГ, ЭхоКГ и 24-часовое холтеровское мониторирование) следует проводить не реже 1 p/год. В случае сердечных аномалий или у пациентов с десминопатиями рекомендуется наблюдение педиатрической кардиологии 2 p/год. Соответственно м.б. введены кардиостимулятор и имплантируемый кардиовертер-дефибриллятор, трансплантация сердца, респираторная поддержка, физиотерапия диапазона движений и вспомогательные устройства. Следует рассмотреть вопрос об исследовании на щелевой лампе для выявления помутнений хрусталика. Известно, что повышенный риск злокачественной гипертермии отсутствует, однако полностью исключить такую возможность пока нельзя.
Генетическое консультирование и пренатальная диагностика должны быть предложены в соответствии с наследственным паттерном и связанным дефектом гена.
Генерация имитирующих пациента клеточных и животных моделей обеспечивает основу для доклинической и клинической оценки новых терапевтических стратегий. Первоначальные исследования на животных основаны на уменьшении интенсивных ФН, лечении антиоксидантом-N-ацетил-L-цистеином, модуляции аутофагической активности и использовании антиагрегационных ЛП, таких как доксициклин и 4-фенилбутират (хим. шаперон, одобренный для лечения нарушений цикла мочевины).