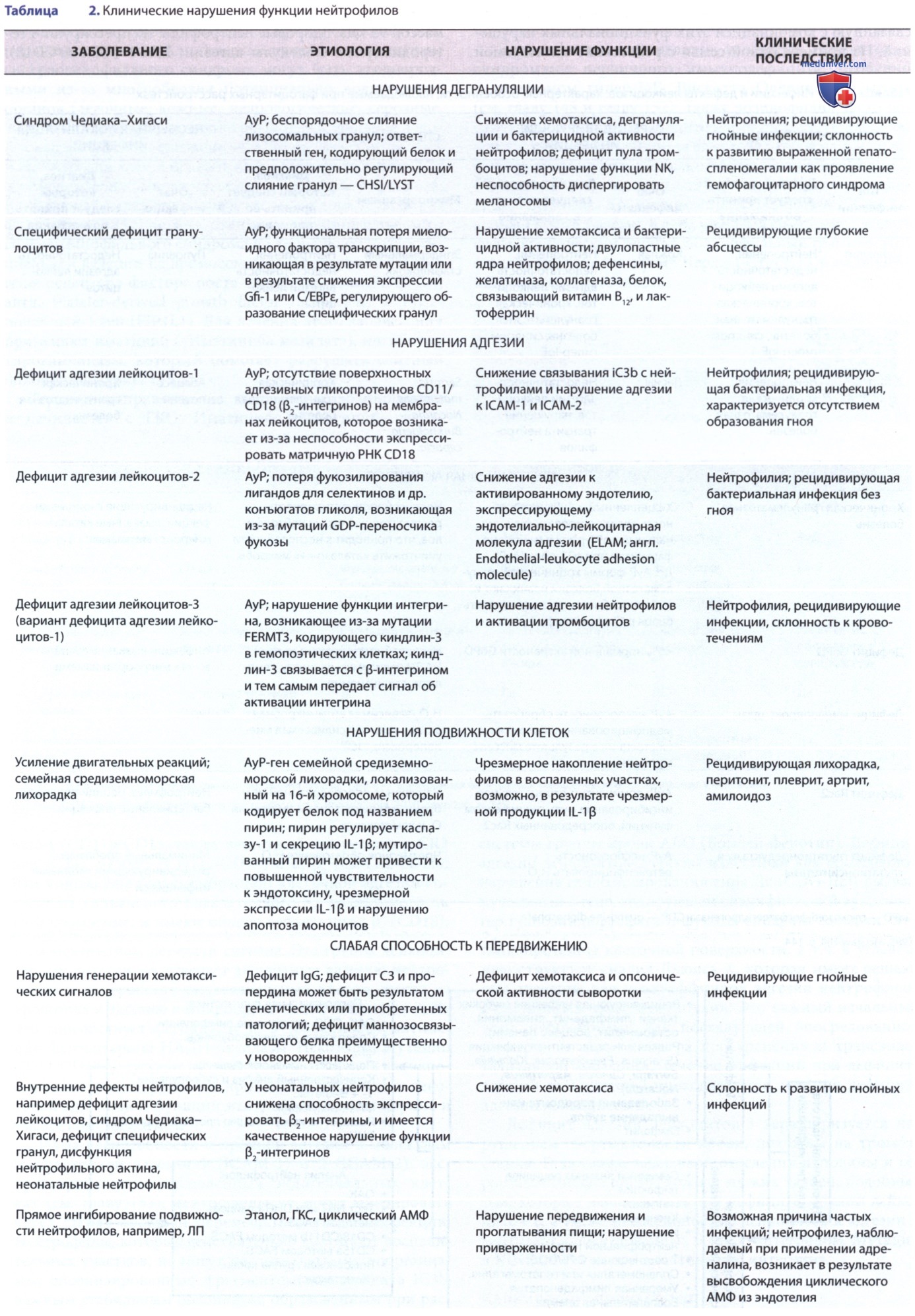
Синдром Чедиака-Хигаси — редкое АуР-заболевание, характеризующееся повышенной восприимчивостью к инфекциям, обусловленной нарушенной дегрануляцией нейтрофилов, легким кровоточащим диатезом, частичным глазокожным альбинизмом, прогрессирующей периферической невропатией и тенденцией к развитию опасной для жизни формы гемофагоцитарного лимфогистиоцитоза. Синдром Чедиака-Хигаси обусловлен базовым дефектом морфогенеза гранул, в результате которого образуются патологически большие гранулы во многих тканях.
Частичное отсутствие пигмента в волосах, коже и глазном дне — результат патологической агрегации меланосом. Неврологическая недостаточность связана с нарушением перекреста зрительного и слухового нервов. Пациенты проявляют повышенную восприимчивость к инфекции, что лишь частично можно объяснить дефектами функции нейтрофилов. У пациентов наблюдается прогрессирующая нейтропения, нарушения функции естественных киллеров (NK), также связанные с дисфункцией гранул.
а) Генетика и патогенез. Ген регулятора лизосомального переноса (LYST), претерпевший обуславливающую синдром Чедиака-Хигаси мутацию, расположен на хромосоме 1q2-q44. Белок LYST регулирует транспорт везикул, опосредуя межбелковое взаимодействие и белково-мембранные ассоциации. Потеря функции может привести к неизбирательному взаимодействию с лизосомными поверхностными белками, в результате чего происходит неконтролируемое слияние лизосом, образуются гигантские гранулы.
Почти во всех клетках пациентов с хроническим сердечным синдромом обнаруживаются дисморфные лизосомы большого размера, накопительные гранулы или связанные с ними везикулярные структуры. Меланосомы имеют слишком большой размер, и их доставка к кератиноцитам и волосяным фолликулам нарушена, в результате чего стержни волос лишены пигментных гранул. Эта аномалия в меланосомах приводит к тому, что волосы и кожа на макроскопическом уровне выглядят светлее, чем ожидалось.
Такая же аномалия в меланоцитах приводит к частичному альбинизму глаз, связанному со светочувствительностью.
Начало развития нейтрофилов, спонтанное слияние гигантских первичных гранул друг с другом или с цитоплазматической мембраной компонентов приводит к образованию огромных вторичных лизосом с пониженным содержанием гидролитических ферментов, включая протеиназы, эластазу и катепсин G. Этот дефицит протеолитических ферментов может быть причиной нарушений нейтрофильной функции уничтожения микроорганизмов при синдроме Чедиака-Хигаси.
б) Клинические проявления. Пациенты с синдромом Чедиака-Хигаси имеют светлую кожу и серебристые волосы, часто жалуются на гиперчувствительность к солнечным лучам и светобоязнь, связанную с вращательным нистагмом. Др. признаки и симптомы значительно различаются, но часто встречаются инфекционные заболевания и невропатии. Инфекции поражают слизистые оболочки, кожу и ДП. Больные дети восприимчивы к гр/п-бактериям, гр/о-бактериям и грибам, причем золотистый стафилококк считается наиболее распространенным возбудителем.
Невропатия может быть сенсорной или моторной, а к характерной особенности относится атаксия. Невропатия часто начинается в подростковом возрасте и становится самой серьезной проблемой.
У пациентов с хроническим сердечным синдромом наблюдается увеличение времени кровотечения при нормальном количестве тромбоцитов в результате нарушения агрегации тромбоцитов, связанного с дефицитом плотных гранул, содержащих аденозиндифосфат и серотонин.
Наиболее опасное для жизни осложнение синдрома Чедиака-Хигаси — развитие ускоренной фазы, характеризующейся панцитопенией, высокой температурой тела (ТТ) и лимфогистиоцитарной инфильтрацией печени, селезенки и лимфатических узлов (ЛУ). Начало ускоренной фазы, которая может произойти в любом возрасте, признано генетической формой гемофагоцитарного лимфогистиоцитоза. Она развивается у 85% пациентов и обычно приводит к летальному исходу.
в) Результаты лабораторных исследований. Диагноз синдрома Чедиака-Хигаси устанавливается при обнаружении крупных включений во всех ядросодержащих клетках крови. Их можно увидеть в мазках крови, окрашенных по Райту: при окрашивании пероксидазой цвет становится более выраженным. Из-за затрудненного выхода из костного мозга клетки, содержащие крупные включения, не выявляются в мазке периферической крови, но легко идентифицируются при исследовании костного мозга. У пациентов наблюдается прогрессирующая нейтропения и нарушение функции тромбоцитов, нейтрофилов и NK-клеток.
г) Лечение. Высокие дозы аскорбиновой кислоты (200 мг/сут для младенцев; 2000 мг/сут для взрослых) могут улучшить клинический статус некоторых детей в стабильной фазе. По поводу эффективности аскорбиновой кислоты существуют разногласия, учитывая безопасность витамина, целесообразно назначать его всем пациентам.
Единственная относительно эффективная терапия для предотвращения ускоренной фазы — трансплантация гемопоэтических стволовых клеток. Нормальные стволовые клетки восстанавливают гематопоэтическую и иммунологическую функцию, корректируют дефицит NK-клеток и предотвращают переход в ускоренную фазу, но не могут исправить или предотвратить невропатию. Если пациент находится в ускоренной фазе с активным гемофагоцитарным лимфогистиоцитозом, трансплантация гемопоэтических стволовых клеток не может предотвратить летальный исход.