Некоторые генетические заболевания наследуются не по классическим менделевским типам. Неменделевское наследование наблюдается при митохондриальных нарушениях, болезнях увеличения триплетных повторов и дефектах импринтинга.
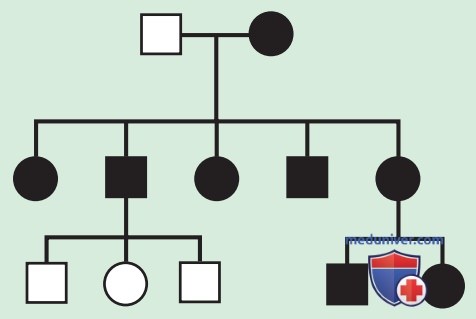
а) Митохондриальное наследование. Митохондриальный геном человека полностью унаследован от матери, поскольку в сперме относительно мало митохондрий, и они разрушаются после оплодотворения. Отсюда следует, что митохондриальное наследование — это, по сути, материнское наследование.
Женщина с митохондриальным генетическим заболеванием может передать его потомству любого пола, но больной отец не может передать болезнь своему потомству (рис. 1). Мутации митохондриальной ДНК часто бывают деле-циями/точечными мутациями; в целом 1 человек из 400 имеет патогенную мутацию митохондриальной ДНК, наследуемую по материнской линии.
В отдельных семьях митохондриальное наследование может быть трудно отличимо от АуД/Х-сцепленного наследования, но во многих случаях пол передающих и не передающих родителей может указывать на митохондриальную природу (табл. 1).
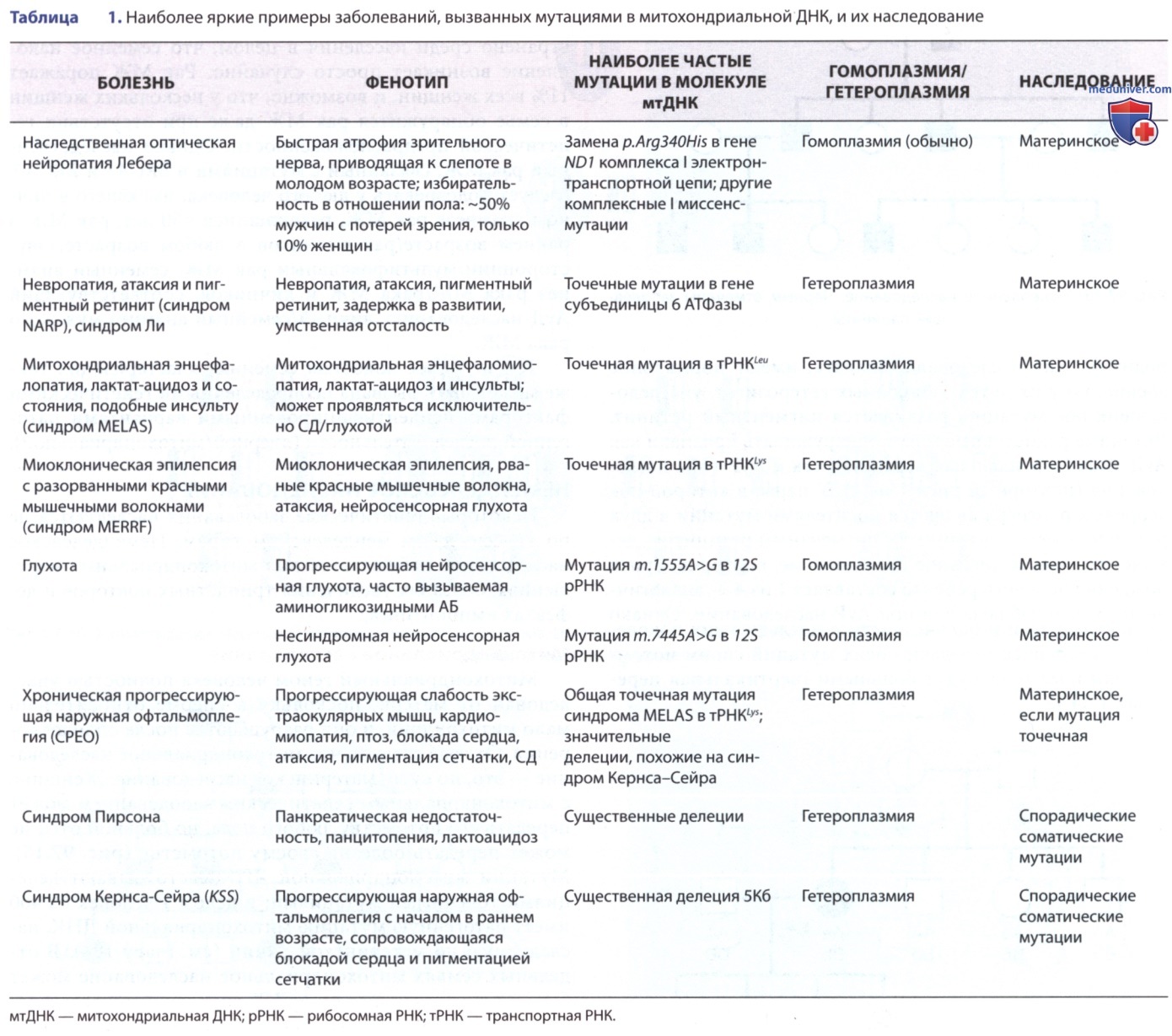
Митохондрии являются поставщиками энергии для клетки, и неудивительно, что органы, на которые больше всего влияет присутствие аномальных митохондрий, — это те органы, которые имеют наибольшую потребность в энергии, такие как ГМ, мышцы, сердце и печень (рис. 2). Общие проявления включают задержку развития, судороги, сердечную дисфункцию, снижение мышечной силы и тонуса, а также проблемы со слухом и зрением.
Рисунок 2. Охват пораженных тканей и клинических фенотипов, связанных с мутациями в митохондриальной ДНК (мтДНК)
Митохондриальные заболевания могут сильно различаться по своим клиническим проявлениям. Отчасти это связано с тем, что клетки обычно содержат несколько митохондрий, каждая из которых несет несколько копий митохондриального генома. Таким образом, в клетках может находиться несколько нормальных и дефектных митохондриальных геномов, что называется гетероплазмией. Напротив, термином «гомоплазмия» определяется состояние, в котором все копии митохондриального генома несут один и тот же вариант последовательности. Неравномерная сегрегация митохондрий, несущих нормальные и дефектные геномы, и репликативное преимущество может привести к различной степени гетероплазмии в клетках заболевшего человека, включая отдельные яйцеклетки больной женщины.
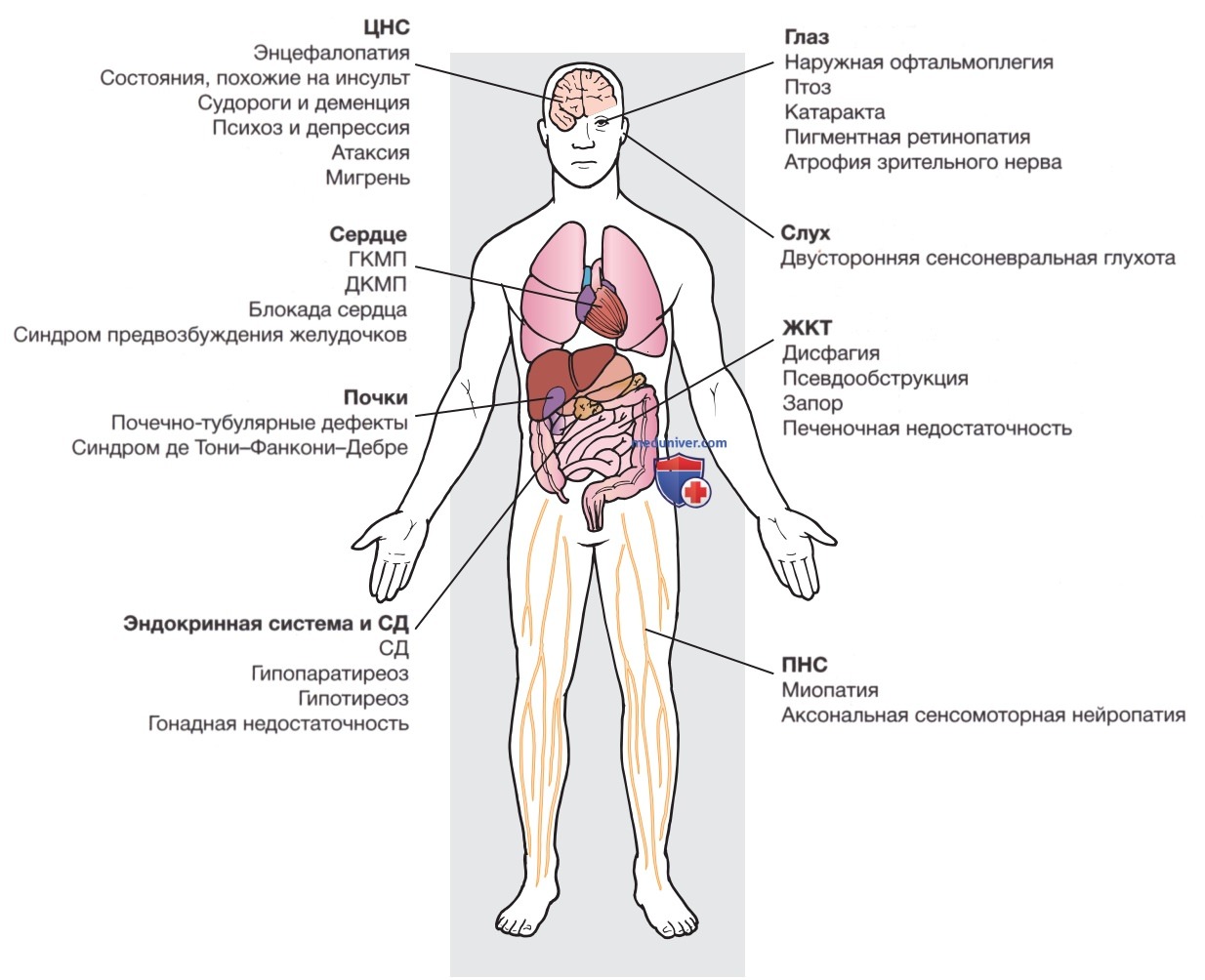
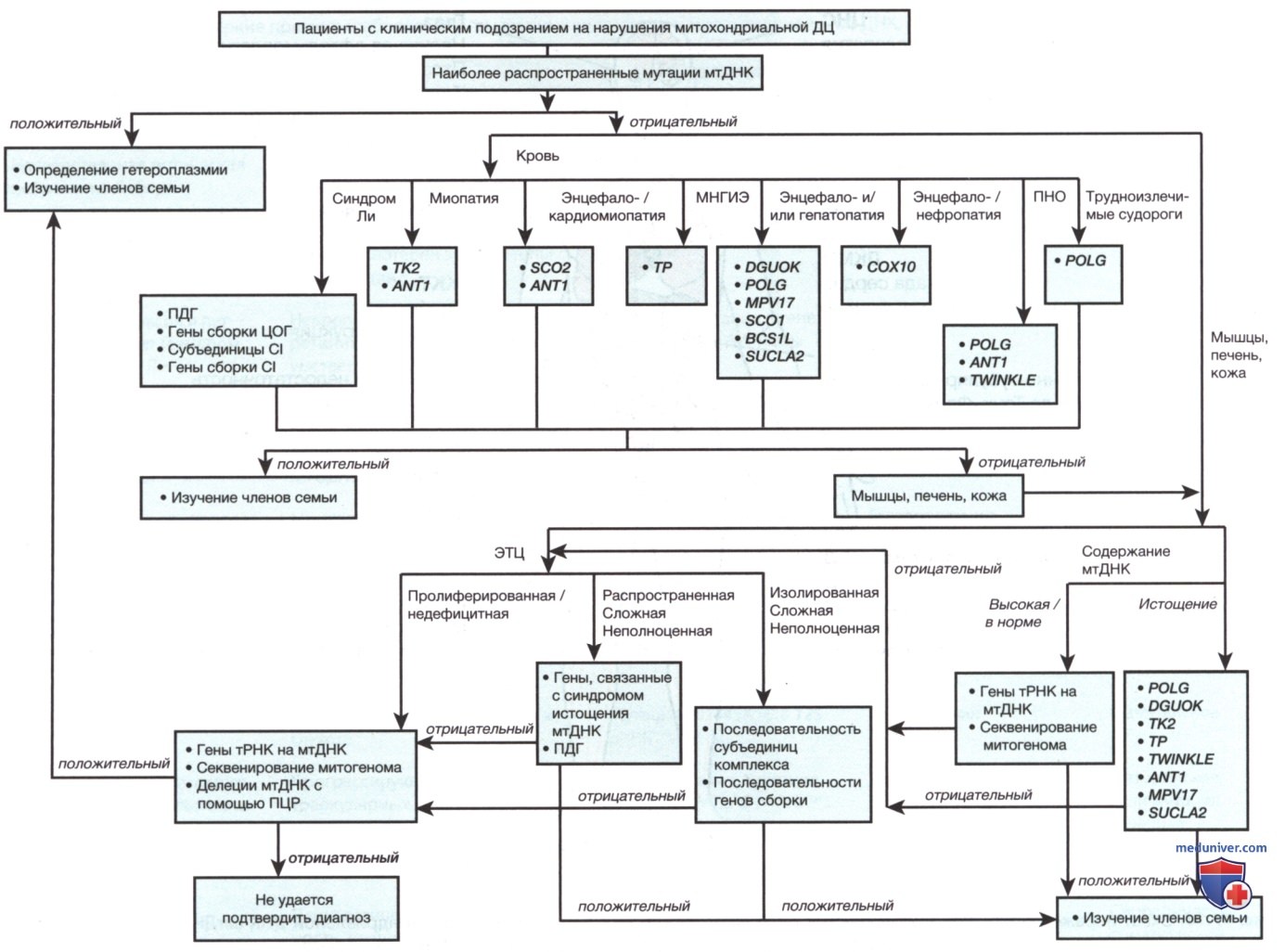
Поэтому заболевание у матери может иметь бессимптомное течение, но у ее детей принимать тяжелые формы. Уровень гетероплазмии, при котором обычно появляются симптомы заболевания, также может варьировать в зависимости от типа митохондриальной мутации. Для обнаружения мутаций митохондриального генома может потребоваться отбор образцов пораженной ткани для последующего анализа ДНК. В некоторых тканях, например крови, тестирование мутаций митохондриальной ДНК может быть недостаточным, поскольку мутация в первую очередь обнаруживается в пораженных тканях, например мышцах (рис. 3).
Рисунок 3. Клинический алгоритм генетического диагностического тестирования генов митохондриальной ДНК (мтДНК) и ядерной ДНК (яДНК) у пациентов с подозрением на митохондриальные нарушения (Baylor College of Medicine, Mitochondrial Diagnostics Laboratory). ДЦ — дыхательная цепь; МНГИЭ — митохондриальная нейрогастроинтестинальная энцефалопатия; ПНО — прогрессирующая наружная офтальмоплегия; ПДГ — пируватдегидрогеназа; CI — комплекс I дыхательной цепи; ЭТЦ — электронная транспортная цепь.
Фактор роста и дифференцировки 15 (GDF-15) и уровни лактата в крови являются скрининговыми тестами для выявления митохондриальных нарушений.
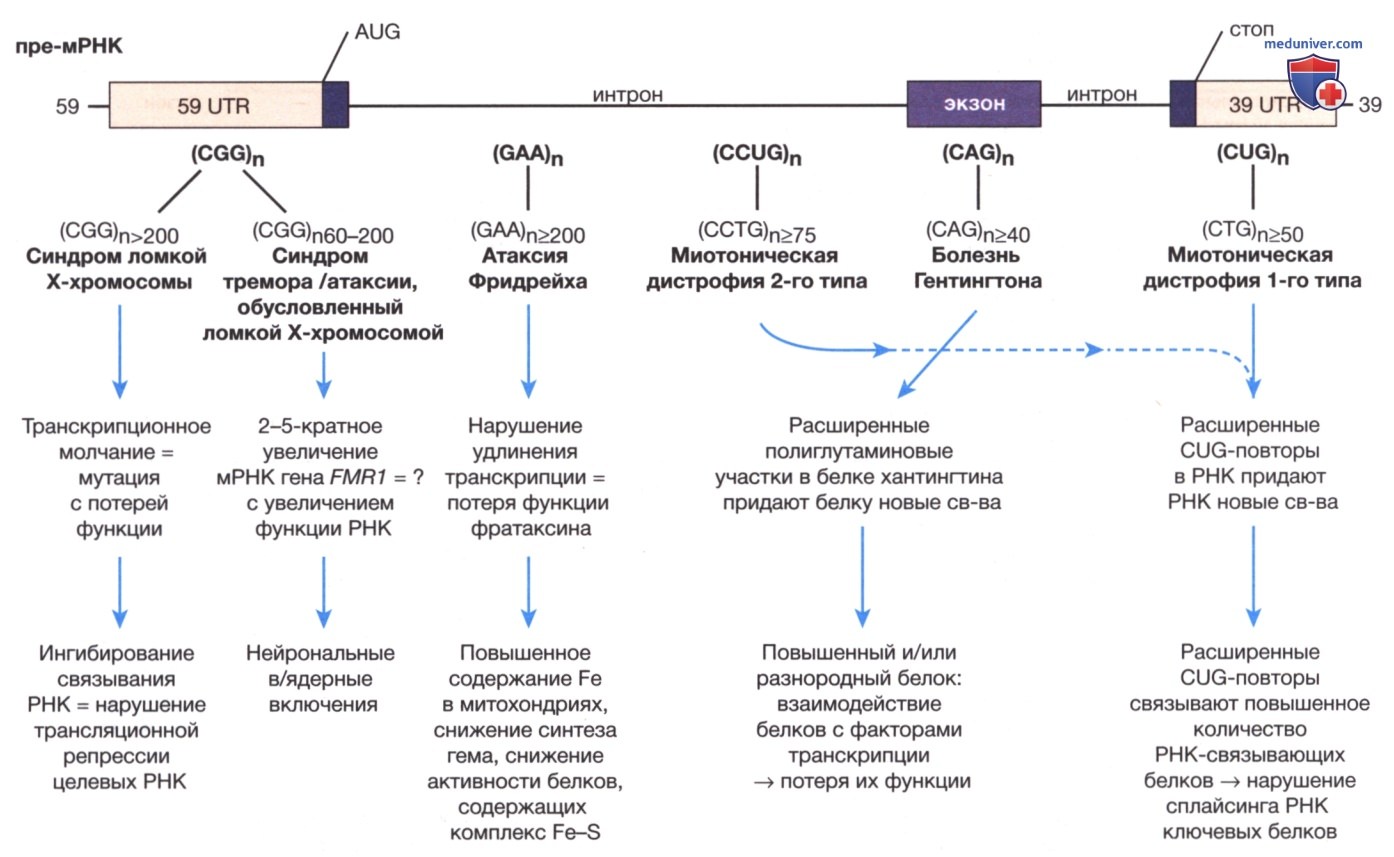
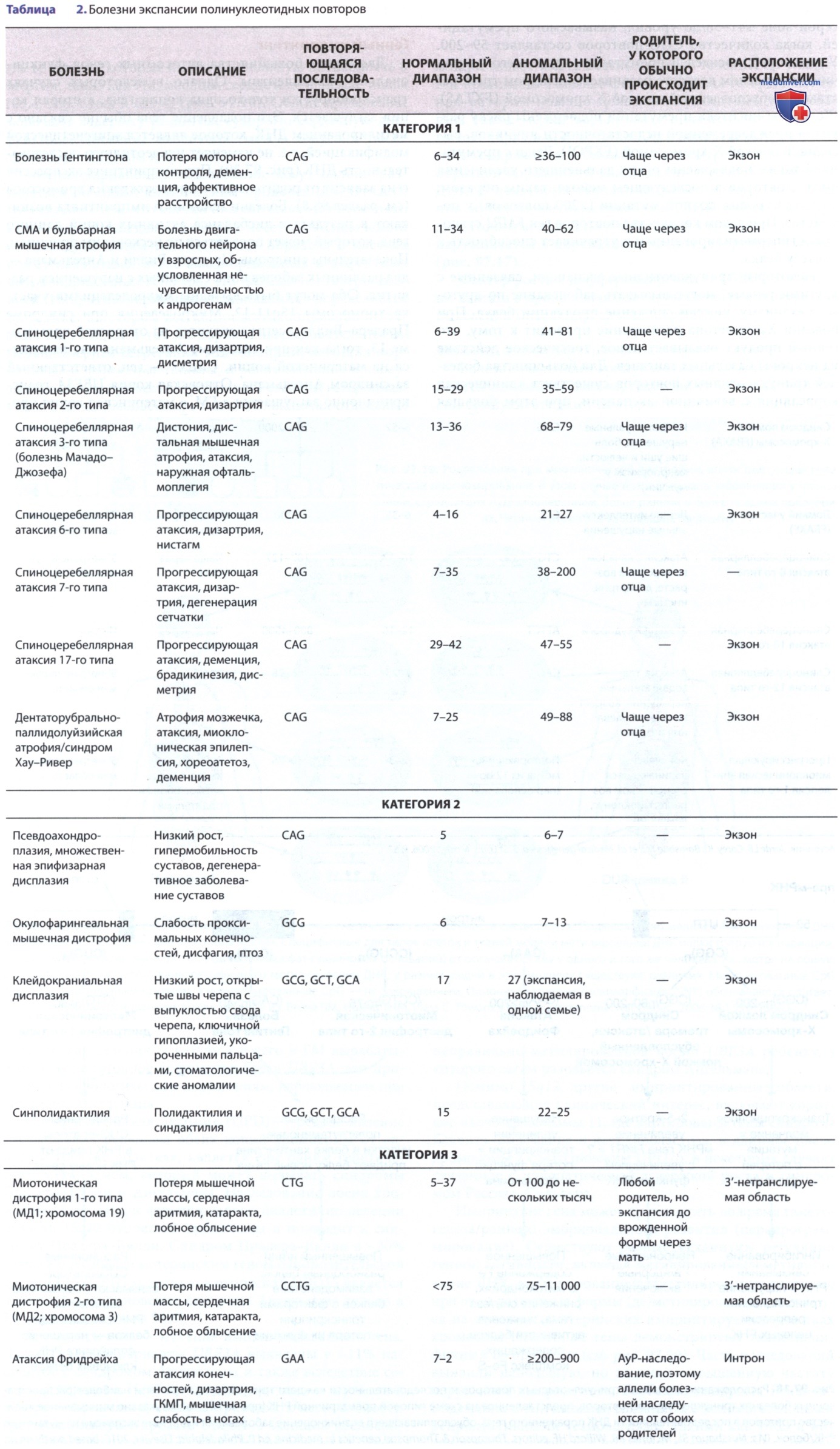
б) Болезни экспансии тринуклеотидных повторов. Болезни экспансии тринуклеотидных повторов вызываются динамическими мутациями. К болезням экспансии тринуклеотидных повторов относятся синдром ломкой Х-хромосомы, миотоническая дистрофия, болезнь Хантингтона и спиноцеребеллярная атаксия (табл. 2 и рис. 4). Эти заболевания вызваны увеличением количества повторов из 3bp (пар оснований). Так, ген FMR1, расположенный на Х-хромосоме, обычно имеет 5-40 тринуклеотидных CGG-повторов.
Рисунок 4. Расположение экспансий тринуклеотидных повторов и последовательности каждого тринуклеотида при пяти наиболее распространенных болезнях тринуклеотидных повторов, представленное на схеме типовой прематричной РНК (пре-мРНК). Также указано минимальное количество повторов в последовательности ДНК пораженного гена, обусловливающего возникновение заболевания и влияние экспансии на мутантную РНК/белок
Ошибка репликации может привести к увеличению числа повторов в серой зоне <41-58/до уровня, называемого премутацией, когда количество CGG-повторов составляет 59-200. У некоторых носителей премутации, чаще всего у мужчин, во взрослом возрасте развивается синдром тремора/ атаксии, обусловленный ломкой Х-хромосомой (FXTAS). Женщины-носители премутации подвержены риску развития преждевременной недостаточности яичников, вызванной ломкой Х-хромосомой (FXPOI). Люди с премутацией также подвержены риску дальнейшего увеличения числа повторов в последующем мейозе, таким образом, достигая уровня полной мутации (>200 повторов) у потомства. При таком количестве повторов ген FMR1 становится гиперметилированным и утрачивает способность к синтезу белка.
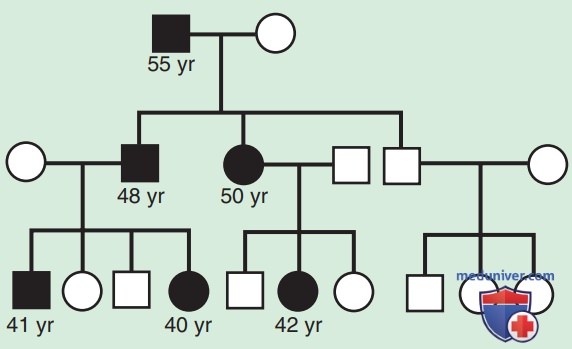
Некоторые тринуклеотидные экспансии, связанные с другими генами, могут вызывать заболевание по другому механизму, нежели снижение продукции белка. При болезни Хантингтона расширение приводит к тому, что генный продукт оказывает новое, токсическое действие на нейроны базальных ганглиев. Для большинства болезней тринуклеотидных повторов существует клиническая корреляция с величиной экспансии, при этом большая экспансия вызывает более тяжелые симптомы и более ранний возраст начала заболевания. Прогрессирующее усиление симптомов наследственного заболевания и снижение возраста начала заболевания в последующих поколениях называется генетической антиципацией и является определяющей характеристикой многих болезней экспансии тринуклеотидных повторов (рис. 5).
Рисунок 5. Родословная при миотонической дистрофии, иллюстрирующая генетическое прогнозирование. В этом случае возраст начала заболевания у членов семьи, страдающих АуД-заболеванием, более ранний в более поздних поколениях. Черным отмечены заболевшие пациенты
в) Генный импринтинг. Две копии большинства аутосомных генов функционально эквивалентны. Однако в некоторых случаях транскрибируется только одна копия гена, а вторая копия заглушается. Это подавление гена обычно связано с метилированием ДНК, которое является эпигенетической модификацией, т.е. не изменяет нуклеотидную последовательность ДНК (рис. 6). При импринтинге экспрессия гена зависит от родительского происхождения хромосомы. Болезни геномного импринтинга возникают в результате дисбаланса активных копий данного гена, который может появляться по нескольким причинам. Показательны синдромы Прадера-Вилли и Ангельмана — два различных заболевания, связанных с нарушением развития. Оба могут быть вызваны микроделециями участка хромосомы 15q11-12.
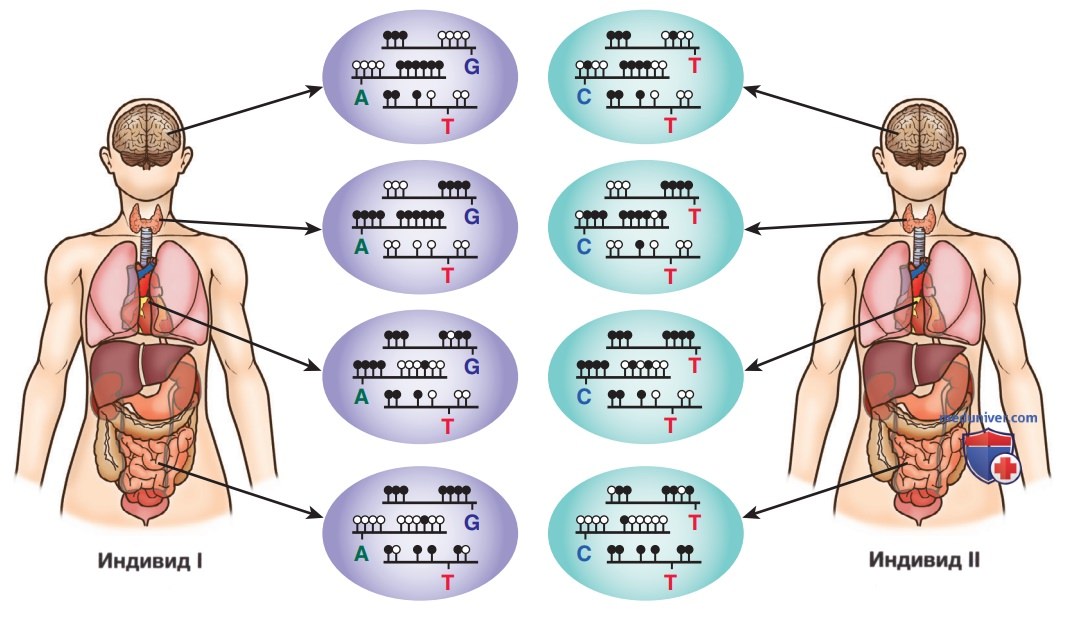
Рисунок 6. Тканеспецифичное метилирование ДНК и эпигенетическая гетерогенность среди людей. Подмножество моделей метилирования ДНК внутри клетки характерно для этого типа клеток. Специфичные для типов клеток и тканей модели метилирования ДНК иллюстрируются вариациями кластеров метилированных CpGs (цитозин-фосфат-гуаниновых оснований) от органа к органу у одного и того же человека. Несмотря на общую согласованность моделей тканеспецифичного метилирования ДНК, у разных людей в этих моделях существуют различия. Метилированные CpG показаны закрашенным кружком, а неметилированные CpG — не закрашенным. Однонуклеотидные полиморфизмы (SNP) обозначаются соответствующим основанием
Микроделеция при синдроме Прадера-Вилли всегда находится на отцовской хромосоме 15, тогда как при синдроме Ангельмана она находится на материнской копии. UBE3A — ген, ответственный за синдром Ангельмана. Отцовская копия UBE3A транскрипционно заглушается в ГМ, а материнская копия продолжает транскрибироваться. Если у человека имеется материнская делеция, в результате чего в ГМ вырабатывается недостаточное количество белка UBE3A, это приводит к неврологическим нарушениям, наблюдаемым при синдроме Ангельмана.
Однородительская дисомия (UPD) — редкое явление наследования ребенком обеих копий хромосомы от одного и того же родителя, является еще одним генетическим механизмом, который может вызывать синдромы Прадера-Вилли и Ангельмана. Наследование обеих хромосом 15 от матери функционально аналогично делеции области 15q12 отцовской хромосомы и приводит к синдрому Прадера-Вилли. Синдром Прадера-Вилли в -30% случаев обусловлен материнским геном однородительской дисомии 15 (UPD15), тогда как отцовский UPD15 является причиной возникновения синдрома Ангельмана только в 3% случаев.
Другая причина — мутация импринтированного гена. Патологические варианты UBE3A выявлены у 11% пациентов с синдромом Ангельмана и также вследствие семейной передачи. Наиболее редкая причина — мутация в центре импринтинга, которая приводит к неспособности правильно импринтировать UBE3A. У женщин неспособность переустановить геномный импринтинг на ее отцовской копии хромосомы 15 вызывает 50% риск передачи неправильно метилированной копии UBE3A ребенку, у которого затем разовьется синдром Ангельмана.
Помимо 15q12, другие импринтированные области, представляющие клинический интерес, включают короткое плечо хромосомы 11, где картированы гены синдрома Беквита-Видеманна и незидиобластоза, и длинное плечо хромосомы 7 с материнским UPD7q, в некоторых случаях связанное с идиопатической задержкой роста и синдромом Рассела-Сильвера.
Импринтинг гена может происходить во время гаметогенеза/раннего эмбрионального развития (перепрограммирование). Существуют разные механизмы регуляции генной активности, включая метилирование/деметилирование ДНК, ацетилирование/деацетилирование гистонов, при этом различные формы (де)метилирования отмечаются на отцовских/материнских импринтируемых участках хромосом. Некоторые гены демонстрируют тканеспецифичный импринтинг (см. рис. 6). Часть исследований выявили небольшую, но значимо повышенную частоту нарушений импринтинга, особенно синдрома Беквита-Видеманна и Ангельмана, связанных с вспомогательными репродуктивными технологиями, такими как ЭКО и интрацитоплазматическая инъекция спермы.
Однако общая частота возникновения этих заболеваний у детей, зачатых с использованием вспомогательных репродуктивных технологий, вероятно, не будет превышать 1%.