Существует три классические формы генетического наследования: АуД-тип, АуР-тип и Х-сцепленный. Их называют менделевскими формами наследования в честь монаха Грегора Менделя, который жил в конце XIX в. и на основании своих опытов сформулировал законы сегрегации признаков, доминирования и независимого наследования. Эти законы лежат в основе закономерностей моногенного наследования.
а) Аутосомно-доминантное наследование. АуД-наследование характеризуется наличием одного патологического гена, расположенного в одной из аутосом (хромосомы 1-22). Аутосомные гены существуют парами, каждый родитель передает ребенку по одной копии. При АуД-типе наследования изменение одного из парных генов влияет на фенотип человека, даже если другая копия гена функционирует правильно. Фенотип может определять особенности физического проявления, поведенческих характеристик, различия, которые можно обнаружить только с помощью лабораторных тестов.
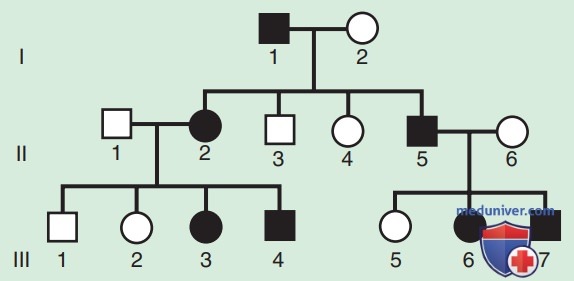
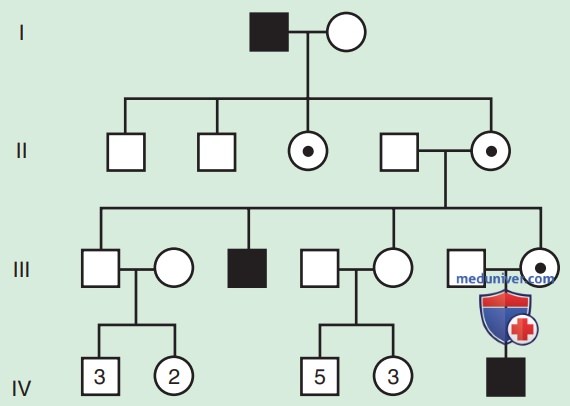
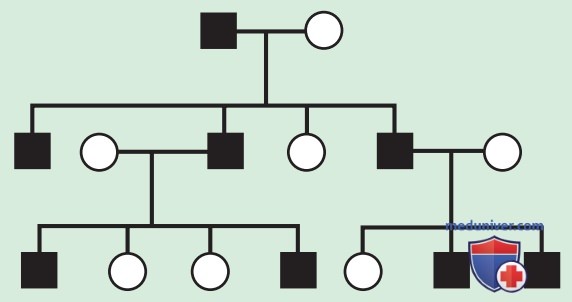
Родословная по АуД-заболеваниям имеет характерные особенности. Эти заболевания распространяются по вертикальному пути передачи (от родителей к ребенку) и могут присутствовать в нескольких поколениях. На рис. 1 это проиллюстрировано тем, что индивид I.1 передает измененный ген индивидам II.2 и II.5. Больной человек имеет один шанс из двух (50% вероятность) передать мутантный ген при каждой беременности и, следовательно, иметь ребенка с этим заболеванием.
Рисунок 1. Родословная по АуД-типу наследования признака. Родословная, показывающая типичное наследование одной из форм хондроплазии (FGFR3), унаследованной как АуД-признак. Черным обозначены заболевшие пациенты
Подобный риск рождения больного ребенка называется риском повторного возникновения заболевания в семье. Не заболевшие лица (члены семьи, которые не проявляют симптомов и не имеют мутантного гена) не передают заболевание своим детям. Заболеванию одинаково подвержены мужчины и женщины.
Хотя сам по себе факт обнаружения передачи от мужчины к мужчине не является определяющим тип наследования признаком, он по существу подтверждает АуД-наследование. Вертикальная передача также может наблюдаться для Х-сцепленных признаков. Однако поскольку отец передает сыну свою Y-хромосому, передачу от мужчины к мужчине нельзя обнаружить по Х-сцепленному признаку. Таким образом, передача от мужчины к мужчине исключает Х-сцепленное наследование как возможное объяснение. Хотя передача от мужчины к мужчине также может происходить по Y-сцепленному типу, существует очень мало У-сцепленных заболеваний по сравнению с тысячами, имеющими АуД-тип наследования.
Хотя передача от родителей ребенку является характерной чертой АуД-наследования, у многих пациентов с АуД-заболеванием нет семейной истории заболевания по нескольким возможным причинам. Во-первых, у пациента может развиться заболевание из-за мутации de novo, которая произошла в ДНК яйцеклетки/сперматозоида, сформировавших его эмбрион.
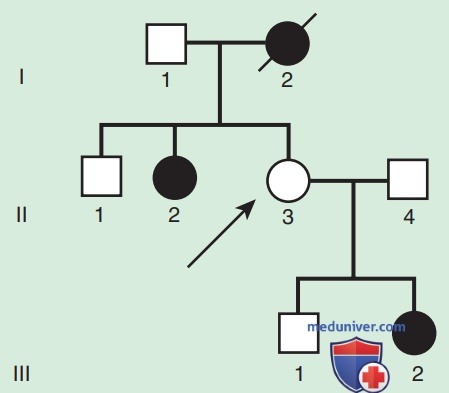
Во-вторых, многие АуД-заболевания характеризуются неполной пенетрантностью, что означает, что не все люди, несущие мутацию, имеют фенотипические проявления. В родословной это может проявляться как пропущенное поколение, в котором незаболевший индивид связан с двумя заболевшими родственниками (рис. 2). Существует множество возможных причин, в связи с которыми заболевание может проявлять неполную пенетрантность, включая влияние генов-модификаторов, факторов окружающей среды, пола и возраста. В-третьих, одно и то же АуД-заболевание у разных людей может иметь симптомы различной степени выраженности.
Рисунок 2. Неполная пенетрантность. Пример семейной сегрегации ракового синдрома и семейного аденоматозного полипоза. Пациент II.3 является облигатным носителем, но данных, свидетельствующих о наличии расстройства, нет. Это заболевание не имеет пенетрантности у этого человека. Черным обозначены заболевшие пациенты
Это обозначается термином «переменная экспрессия» и является характерной особенностью многих АуД-заболеваний. В-четвертых, некоторые спонтанные генетические мутации происходят не в яйцеклетке/сперматозоиде, из которых формируется ребенок, а в клетке развивающегося эмбриона. Такие трансформации называются соматическими мутациями и, поскольку они не затрагивают все клетки, считаются мозаичными. Фенотип, вызванный соматической мутацией, может варьировать, но обычно он более мягкий, чем если бы все клетки были затронуты мутацией. При гонадном мозаицизме мутация происходит в клетках зародышевой линии, ведущих к образованию ооцитов/сперматозоидов.
Человек с гонадным мозаицизмом может не иметь никаких проявлений заболевания, но производить яйцеклетки/спермато-зоиды, затронутые мутацией.
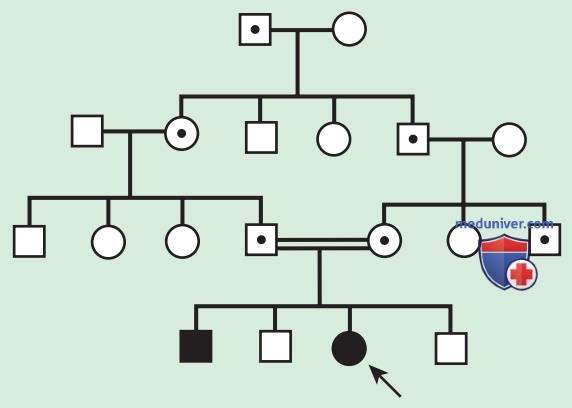
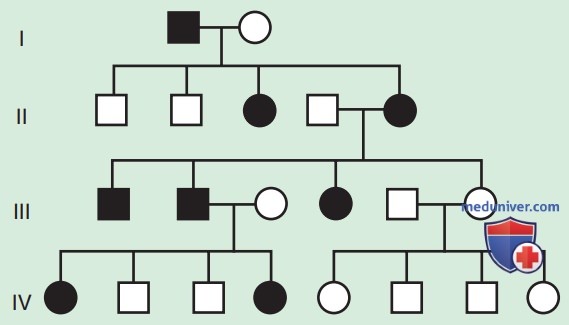
б) Аутосомно-рецессивное наследование. АуР-наследование заболеваний — тип наследования, при котором генетически обусловленная болезнь проявляется только при наличии патологических мутаций в обеих копиях гена. Примерами являются муковисцидоз и СКА. АуР-заболевания характеризуются горизонтальной передачей, наличием нескольких заболевших членов семьи в одном поколении, но не заболевших членов семьи в других поколениях (рис. 3).
Рисунок 3. Родословная по АуР-типу наследования признака в случае кровного родства родителей. Точкой в центре отмечены носители. Черным — заболевшие пациенты
Для родителей-носителей, у которых ранее был рожден больной ребенок, они связаны с 25% риском повторного возникновения заболевания в семье.
Потомки мужского и женского пола могут заболеть с одинаковой вероятностью, хотя некоторые признаки проявляются по-разному для каждого пола. Потомки родителей, связанных между собой кровным родством, подвергаются повышенному риску возникновения редких АуР-признаков из-за повышенной вероятности наследования гена с патологической мутацией от обоих родителей, который они, в свою очередь, унаследовали от общего предка. Кровное родство между родителями ребенка с подозрением на генетическое заболевание позволяет заподозрить АуР-наследование, но не доказывает его.
Хотя кровнородственные союзы необычны в западном обществе, в других частях мира (Южная Индия, Япония и Ближний Восток) <50% всех детей могут быть зачаты в кровных союзах. Риск возникновения генетического заболевания для потомков от брака между двоюродными родственниками (6-8%) в ~2 раза выше, чем в общей группе (3-4%).
У каждого человека, по всей вероятности, есть несколько редких патогенных рецессивных мутаций. Поскольку частота возникновения большинства патогенных мутаций в основной массе населения очень низка, проверять все население для выявления небольшого числа лиц-носителей этих мутаций не имеет экономического смысла. В результате эти варианты обычно остаются необнаруженными, если только заболевший ребенок не рождается от семейной пары, в которой оба супруга являются носителями патогенной мутации, влияющей на один и тот же ген.
Однако в некоторых генетических изолятах (небольшие группы лиц, изолированные по географическому, религиозному, культурному, языковому признаку) отдельные редкие рецессивные патогенные варианты встречаются гораздо чаще, чем у основной массы населения. Несмотря на то что родство может быть неизвестным, супружеские пары из этих генетических изолятов получают больше шансов иметь общие патогенные аллели, унаследованные от общего предка. Среди некоторых таких групп были разработаны программы скрининга для выявления лиц, являющихся носителями наиболее распространенных болезнетворных вариантов и, следовательно, подверженных повышенному риску передать их своим детям.
Случаи разнообразных АуР-заболеваний чаще всего наблюдаются среди евреев-ашкенази, чем среди населения в целом. Еврейским парам ашкеназского происхождения следует предложить пренатальный скрининг/скрининг до зачатия на болезнь Гоше 1-го типа (частота носителей 1:14), муковисцидоз (1:25), болезнь Тея-Сакса (1:25), семейную дизавтономию (1:30), болезнь Канавана (1:40), болезнь накопления гликогена типа 1А (1:71), лейциноз (1:81), анемию Фанкони типа С (1:89), болезнь Ниманна-Пика типа А (1:90), синдром Блума (1:100), муколипидоз IV (1:120) и, возможно, семейную гиперинсулинемическую гипогликемию младенцев.
Распространенность носительства некоторых АуР-вариантов в более крупных группах необычно высока. В таких случаях постулируется гетерозиготное преимущество. Частота носительства СКА среди африканского населения и муковисцидоза среди североевропейской этнической группы намного выше, чем можно было бы ожидать, исходя из частоты новых мутаций. В этих популяциях гетерозиготные носители могут иметь преимущество с точки зрения выживания и воспроизводства по сравнению с не носителями.
Считается, что при СКА состояние носительства придает некоторую устойчивость к малярии; постулируется, что при муковисцидозе состояние носительства обеспечивает устойчивость к холере/ энтеропатогенным инфекциям, вызываемым Escherichia coli. Для лиц североевропейского и еврейского происхождения ашкенази рекомендуется популяционный скрининг на носительство муковисцидоза; лицам африканского происхождения — обследование населения на СКА.
Если частота возникновения АуР-заболевания известна, частота гетерозиготного заболевания/носительства может быть рассчитана по формуле Харди-Вайнберга:
р2 + 2pq + q2 = 1,
где р — частота наследования одного аллеля из пары, а q — частота наследования другого.
Например, если частота муковисцидоза среди американцев белой расы составляет 1:2500 (р2), то можно рассчитать частоту гетерозиготы (2pq): если р2 = 1/2500, то р = 1/50 и q = 49/50; 2pq = 2 х х (1/50) х (49/50) = 98/2500, или 3,92%.
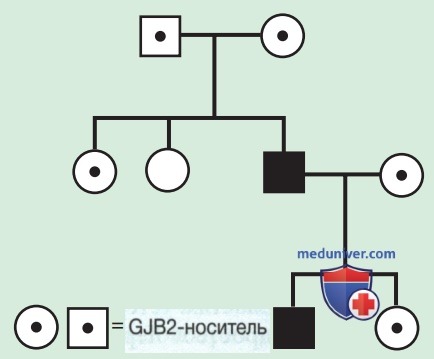
в) Псевдодоминантное наследование. Псевдодоминантное наследование — ситуация выявления очевидной доминантной (от родителя к ребенку) передачи известного АуР-заболевания (рис. 4), происходящая, когда у гомозиготного больного человека есть партнер, являющийся гетерозиготным носителем. Чаще всего это происходит в отношении таких распространенных в группе населения рецессивных признаков, как СКА/ несиндромный АуР-тип потери слуха, вызванные вредными мутациями в GJB2 — гене, кодирующем коннексин 26.
Рисунок 4. Псевдодоминантное наследование. Черным цветом обозначен заболевший (глухотой) пациент; точкой в центре указывают бессимптомного носителя (здорового)
г) Х-сцепленное наследование. Х-сцепленное наследование описывает модели наследования большинства заболеваний, вызванных вредоносными изменениями в генах, расположенных на Х-хромосоме (рис. 5). Х-сцепленные заболевания чаще проявляются у мужчин, чем у женщин. Женщины-носители этих заболеваний, как правило, не больны сами/болеют в гораздо более легкой форме, чем мужчины. При каждой беременности женщины-носительницы имеют 25% шанс родить больного сына, 25% вероятность родить дочь-носительницу и 50% вероятность родить ребенка, не унаследовавшего мутировавший Х-сцепленный ген.
Рисунок 5. Родословная, иллюстрирующая Х-сцепленное рецессивное наследование. Точкой в центре отмечены носители. Черным — заболевшие пациенты
Поскольку заболевшие мужчины передают свою Х-хромосому всем своим дочерям и свою Y-хромосому всем своим сыновьям, у них есть 50% шанс иметь здорового сына, не несущего ген болезни, и 50% шанс иметь дочь-носителя. Передача от мужчины к мужчине исключает Х-сцепленное наследование, но наблюдается при АуД-типе и Y-сцепленном наследовании.
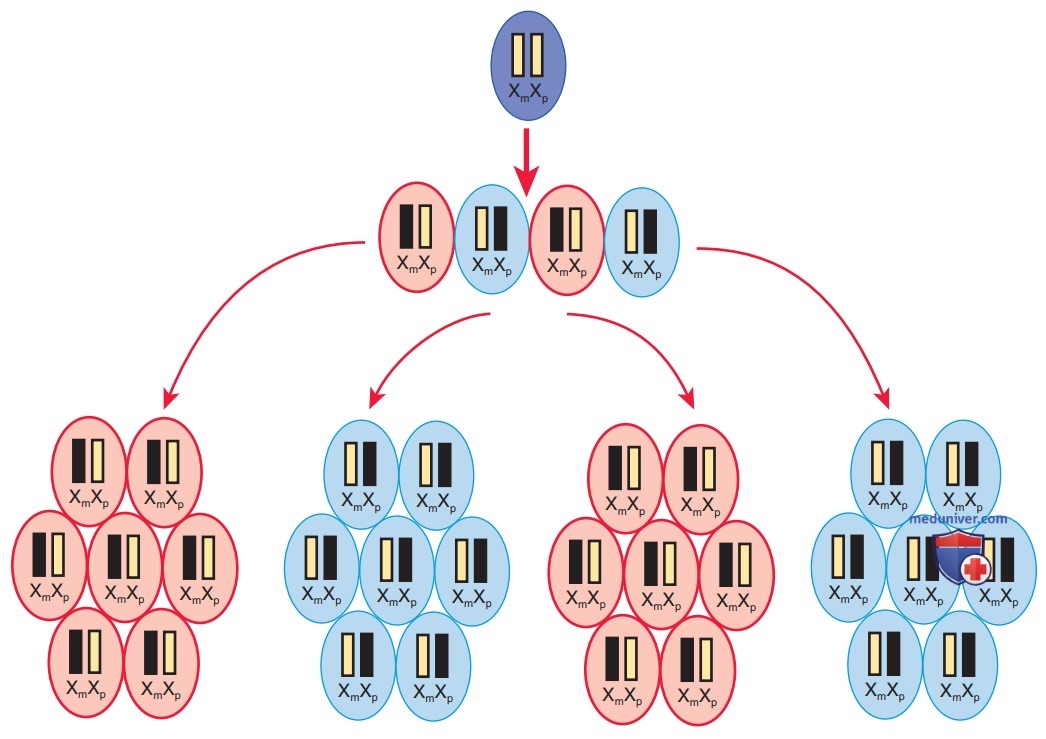
У женщин иногда могут проявляться симптомы Х-сцепленного признака, похожие на таковые у мужчин. Случается это редко вследствие гомозиготности по Х-сцепленному признаку/наличия аномалии половых хромосом (45,X/46,XY, женщина), а также искаженной/неслучайной Х-инактивации. Инактивация Х-хромосомы происходит на ранней стадии развития и состоит в случайной и необратимой инактивации большинства генов на одной Х-хромосоме в женских клетках (рис. 6). В некоторых случаях преобладание клеток инактивирует эту же Х-хромосому, что приводит к фенотипической экспрессии Х-сцепленного патогенного варианта, если он находится на активной хромосоме.
Рисунок 6. Х-инактивация. Черным отмечена активная Х-хромосома. Цвет клетки показывает, что ее активная Х-хромосома происходит от отца (Хp, синий)/от матери (Хm, розовый)
Это может произойти случайно, из-за селективного отбора против клеток, которые инактивировали Х-хромосому, несущую нормальный ген/аномалии Х-хромосомы, которая приводит к инактивации Х-хромосомы, несущей нормальный ген.
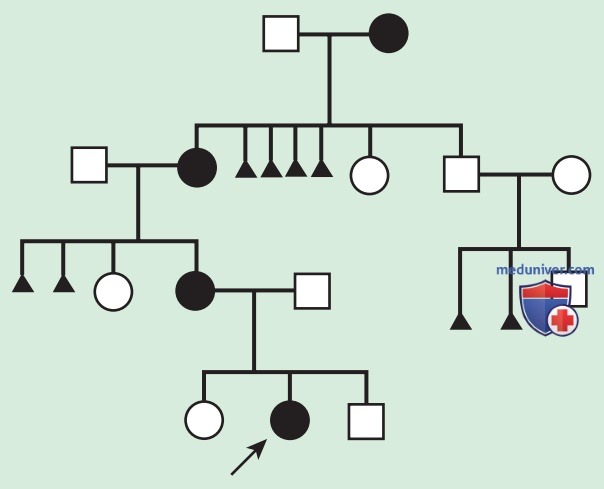
При некоторых Х-сцепленных заболеваниях как гемизиготные мужчины, так и гетерозиготные женщины, несущие дефектный Х-сцепленный ген, имеют сходные фенотипические проявления. В этих случаях у заболевшего мужчины есть 50% шанс иметь больную дочь и 50% шанс иметь здорового сына (при каждой беременности), в то время как половина мужского и женского потомства больной женщины будут больны (рис. 7). Некоторые болезни, связанные с Х-хромосомой, являются летальными для высокого процента мужчин, например пигментная инконтиненция. В таких случаях в родословной обычно указываются только больные женщины и общее соотношение женщин и мужчин (2:1) с увеличением количества выкидышей (рис. 8).
Рисунок 7. Родословная, иллюстрирующая Х-сцепленное доминантное наследование. Черным отмечены заболевшие пациенты. Обратите внимание, что в этой ситуации нет передачи от отца к сыну, и гемизиготность (т.е. ген, сцепленный с Х-хромосомой у мужчин) не является летальной. При некоторых Х-сцепленных доминантных заболеваниях Х-сцепленные мужчины имеют более тяжелый фенотип и могут не выжить. В этом случае заболевание проявляется только у женщин (рис. 12)
Рисунок 8. Родословная при Х-сцепленном доминантном заболевании со смертельным исходом для мужчин (недержание пигмента). Черным отмечены заболевшие пациенты
д) Y-сцепленное наследование. Y-сцепленных признаков существует немного. Они передаются только от мужчины к мужчине и затрагивают только мужчин (рис. 9). Большинство Y-сцепленных генов связано с определением мужского пола и репродукцией, что вызывает бесплодие. Таким образом, передача Y-сцепленного заболевания внутри семьи встречается редко. Однако достижения в области вспомогательных репродуктивных технологий могут сделать возможной семейную передачу мужского бесплодия.
е) Наследование, связанное с псевдоаутосомными регионами. Особого внимания заслуживают псевдоаутосомные регионы на X-/Y-хромосомах. Поскольку эти области состоят из гомологичных последовательностей нуклеотидов, гены, расположенные в этих областях, присутствуют в равном количестве как у мужчин, так и у женщин. SHOX — один из наиболее описанных генов болезней, расположенных в этих областях. Гетерозиготные мутации SHOX вызывают дисхондростеоз Лери-Вейлля — редкую скелетную дисплазию, которая включает двустороннее искривление предплечий с дислокацией локтевой кости к запястью и общий низкий рост. Гомозиготные мутации SHOX вызывают гораздо более тяжелую мезомелическую дисплазию Лангера.