Глюкоза играет центральную роль в энергетическом обмене, экономии энергии и является источником хранения энергии в виде гликогена, жира и белка. В качестве непосредственного источника энергии глюкоза обеспечивает 38 моль АТФ на 1 моль окисленной глюкозы. Глюкоза необходима для энергетического обмена в ГМ, где она обычно является предпочтительным субстратом и на ее освоение приходится почти все потребление кислорода ГМ. ГМ поглощает глюкозу с помощью инсулиннезависимых переносчиков. Транспорт глюкозы в ГМ осуществляется через проницаемые для жидких мембран транспортеры глюкозы в эритроцитах (GLUT-1; англ. Glucose transporter 1) и относится к процессу облегченной диффузии, который зависит от концентрации глюкозы в крови и не регулируется инсулином. Следовательно, низкие концентрации глюкозы в крови приводят к гипогликемии ГМ. Дефицит транспортеров глюкозы в ГМ может привести к судорогам из-за низких концентраций глюкозы в ГМ и СМЖ (гипогликоррахия), несмотря на нормальный уровень глюкозы в крови.
Для поддержания концентрации глюкозы в крови и предотвращения ее резкого падения до уровней, нарушающих функцию ГМ, организмом используется сложная система регулирования гликемии.
Защита от гипогликемии включает ВНС и гормоны, которые действуют согласованно, повышая выработку глюкозы за счет ферментативной модуляции гликогенолиза и глюконеогенеза, одновременно ограничивая периферическую утилизацию глюкозы, которая сохраняет глюкозу для метаболизма ГМ. Гипогликемия представляет собой дефект одного или нескольких сложных взаимодействий, которые обычно интегрируют гомеостаз глюкозы во время приема пищи и голодания. Этот процесс особенно важен для новорожденных, у которых происходит резкий переход от в/утробной жизни, характеризующейся зависимостью от трансплацентарного поступления глюкозы, к самостоятельной, с автономной поддержкой эугликемии. Поскольку недоношенность или плацентарная недостаточность могут ограничивать запасы питательных в-в в организме, а генетические нарушения в ферментных или гормональных системах проявляются быстро, гипогликемия достаточно часто встречается в неонатальном периоде.
а) Определение. У новорожденных не всегда существует очевидная корреляция между концентрацией глюкозы в крови и классическими клиническими проявлениями гипогликемии. Отсутствие симптомов не является гарантией того, что концентрация глюкозы в норме и на безопасном оптимальном уровне для поддержания метаболизма ГМ. Есть данные о том, что гипоксемия и ишемия могут усиливать роль гипогликемии в развитии хронических нарушений ЦНС и возникновении необратимых последствий для ГМ. Нижний предел допустимой нормы уровня глюкозы в крови у новорожденных с сопутствующим заболеванием, которое уже нарушает метаболизм ГМ, не определен. Из-за беспокойства о возможных неврологических, интеллектуальных или психологических последствиях в более позднем возрастном периоде большинство специалистов считают опасным уровень глюкозы в крови <55 мг/дл у новорожденных, настаивая на коррекции. Это особенно важно в первые 2-3 ч жизни, когда глюкоза достигает своего минимального уровня; впоследствии уровень глюкозы в крови начинает повышаться и через 12-24 ч достигает значений >55-65 мг/дл.
К третьему дню жизни у нормальных доношенных новорожденных уровень глюкозы в крови в среднем составляет 65 мг/дл (диапазон 65-100).
Следовательно, у здоровых доношенных новорожденных после третьего дня жизни, а также у младенцев старшего возраста и детей концентрация глюкозы в цельной крови <55 мг/дл (550 мг/л) (на 10-15% выше для сыворотки крови или плазмы) рассматривается как гипогликемия, поскольку при таких концентрациях глюкозы активируются антагонистичные механизмы. У детей старшего возраста определение гипогликемии основано на «триаде Уиппла»: концентрация глюкозы в плазме <60 мг/дл вместе с сопутствующими симптомами, связанными с ЦНС или катехоламинами, а также регрессией симптомов, когда концентрация глюкозы восстанавливается до нормального значения путем коррекции глюкозой.
б) Значение и последствия. Большая часть эндогенной продукции глюкозы в печени у младенцев и детей раннего возраста, наблюдающаяся через несколько часов после кормления и во время голодания, м.б. объяснена ее участием в метаболизме ГМ. Кроме того, в любом возрасте существует корреляция между продукцией глюкозы и массой ГМ.
Поскольку ГМ растет наиболее быстро в первый год жизни и большая часть оборота глюкозы используется для метаболизма ГМ, устойчивая или повторяющаяся гипогликемия у младенцев и детей может замедлить развитие и функционирование ГМ. Преходящая изолированная и бессимптомная краткосрочная гипогликемия, судя по всему, не связана с этими тяжелыми последствиями. В быстро растущем мозге глюкоза также является источником мембранных липидов и обеспечивает синтез структурных белков и миелинизацию, важные для нормального созревания ГМ. В условиях тяжелой и устойчивой гипогликемии эти структурные субстраты ГМ распадаются до энергозатратных промежуточных продуктов, таких как лактат, пируват, аминокислоты и кетокислоты, которые могут поддерживать метаболизм ГМ за счет его роста. Способность ГМ новорожденного поглощать и окислять кетоновые тела в ~5 раз больше, чем у ГМ взрослого. Однако способность печени вырабатывать кетоновые тела в периоде новорожденности ограничена, особенно при наличии гиперинсулинемии, когда подавляются поступление глюкозы из печени в кровь, липолиз и кетогенез, тем самым лишая ГМ любых альтернативных источников энергии.
Хотя ГМ и может метаболизировать кетоны, эти альтернативные виды энергии неспособны полностью заменить глюкозу как основной источник энергии для ЦНС. Недостаток основного источника энергии ГМ во время гипогликемии и ограниченная доступность альтернативных источников энергии во время гиперинсулинемии имеют предсказуемые неблагоприятные последствия для метаболизма и роста ГМ: падение потребления кислорода ГМ и разрушение эндогенных структурных компонентов с нарушением функциональной целостности клеточных мембран. Гипоксия усиливает повреждающее действие гипогликемии, а в условиях даже незначительного снижения уровня глюкозы в крови сама является причиной повреждения ГМ.
Основными долгосрочными последствиями тяжелой продолжительной гипогликемии являются когнитивные нарушения, периодические судороги, церебральный паралич и вегетативная дисфункция. Возможны также незначительные воздействия на личность, но они не были четко определены. К необратимым изменениям в ЦНС у 25-50% пациентов в возрасте до 6 мес (период наиболее быстрого роста мозга) приводят приступы с тяжелой рецидивирующей симптоматической гипогликемией. Они могут сказываться на патологических изменениях, характеризующихся нарушением миелинизации белого в-ва ГМ и атрофией коры больших полушарий, что отражается в увеличении бороздок и сглаживании извилин ГМ. Тяжелые последствия наиболее вероятны, когда альтернативные источники энергии ограничены, как это происходит при гиперинсулинемии, когда эпизоды гипогликемии являются повторяющимися или продолжительными, и когда они усугубляются гипоксией.
Нет точных сведений о предсказуемой связи продолжительности или тяжести гипогликемии с последующим неврологическим развитием детей. Хотя гипогликемия у детей старшего возраста встречается реже, она также может вызывать необратимые повреждения НС из-за опосредованной гибели нейронов, индуцируя выделение нейротоксических медиаторов, выделяемых во время гипогликемии.
в) Субстрат, фермент и гормональная интеграция гомеостаза глюкозы:
1. Новорожденные. В норме глюкоза плода полностью поступает от матери через плаценту. Т.о., концентрация глюкозы у плода соответствует ее уровню, но немного ниже, чем у матери. Высвобождение катехоламинов, происходящее при гипоксии (стрессе) плода, мобилизует глюкозу и СЖК через β-адренергические механизмы, отражая β-адренергическую активность в печени и жировой ткани плода. Катехоламины одновременно ингибируют фетальный инсулин и стимулируют секрецию глюкагона.
Острое прерывание поступления материнской глюкозы плоду во время родов требует необходимость немедленной мобилизации эндогенной глюкозы. Этому способствуют три связанных события: изменения секреции гормонов, их рецепторов и активности ключевых ферментов. В течение нескольких минут или часов после рождения происходит резкое увеличение концентрации глюкагона в 3-5 раз. Уровень инсулина обычно снижается и остается в пределах нижнего диапазона в течение нескольких дней, не демонстрируя ответной быстрой реакции на глюкозу. Также характерен резкий всплеск спонтанной секреции катехоламинов. Адреналин, в свою очередь, увеличивает секрецию СТГ за счет а-адренергических механизмов; уровни СТГ при рождении заметно повышены. Кроме того, уровни кортизола в ближайшем послеродовом периоде выше при родах естественным путем, чем путем кесарева сечения, что частично отражает нагрузку на секрецию в/утробного кортизола. Действуя согласованно, эти гормональные изменения при рождении мобилизуют глюкозу за счет гликогенолиза и глюконеогенеза, активируют липолиз и способствуют кетогенезу.
В результате этих процессов концентрация глюкозы в плазме после кратковременного снижения после рождения стабилизируется, запасы гликогена в печени быстро истощаются и в течение нескольких часов после рождения ~10% оборота глюкозы у новорожденного образуется через глюконеогенез из основной глюконеоген-ной аминокислоты — аланина. Концентрации СЖК также резко увеличивается вместе с резкими скачками уровней глюкагона и адреналина, за которыми позднее следует повышение уровня кетоновых тел. Т.о., глюкоза частично сохраняется для использования ГМ, в то время как СЖК и кетоны являются альтернативными источниками энергии для мышц, а также необходимыми глюконеогенными факторами, необходимыми для управления глюконеогенезом, такими как КоА и НАДН, в результате окисления жирных кислот в печени.
В раннем постнатальном периоде ПЖЖ способствуют усиленной секреции глюкагона для поддержания нормальной концентрации глюкозы в крови. Такие адаптивные изменения в секреции гормонов сопровождаются столь же поразительными адаптивными изменениями в рецепторах гормонов. Ключевые ферменты, участвующие в производстве глюкозы, также резко меняются в перинатальном периоде. Т.о., происходит быстрое падение активности гликогенсинтазы и резкое повышение активности фосфорилазы. Резко повышается после рождения активность фермента, ограничивающего скорость глюконеогенеза, фосфоенолпируваткарбоксикиназы, частично активируясь при повышении уровня глюкагона и снижении уровня инсулина. Данная закономерность может объяснить несколько причин гипогликемии новорожденных, основанных на нарушении гормональной секреции и дефицитом субстратов, в виде гликогена печени, мышц, как источника аминокислот для глюконеогенеза, и запасов липидов, для высвобождения жирных кислот. Или недостаточностью ключевых ферментов, регулирующих гомеостаз глюкозы (см. рис. 1).
2. Младенцы старшего возраста и дети. Гипогликемия у младенцев старшего возраста и детей аналогична гипогликемии у взрослых, у которых гомеостаз глюкозы, сразу после еды поддерживается за счет гликогенолиза и глюконеогенеза — в интервале между приемами пищи. В печени ребенка с МТ 10 кг содержится 20-25 г гликогена, и этого достаточно для удовлетворения нормальных потребностей в глюкозе в 4-6мг/кг в минуту в течение 6-12 ч. По истечении этого периода должен активироваться печеночный глюконеогенез. И гликогенолиз, и глюконеогенез зависят от метаболических путей, представленных на рис. выше. Нарушения гликогенолиза или глюконеогенеза могут не проявляться у младенцев до тех пор, пока их кормят с интервалами 3-4 ч, и проявляются, когда их перестают кормить по ночам, что обычно наблюдается к 3-6 мес жизни. Источником глюконеоген-ных предшественников является прежде всего мышечный белок. Мышечная масса младенцев и маленьких детей значительно меньше МТ взрослых, тогда как потребность в глюкозе на единицу МТ у детей выше. Следовательно, способность компенсировать недостаток глюкозы за счет глюконеогенеза более ограничена у младенцев и детей младшего возраста, как и способность выдерживать голодание в течение продолжительных периодов времени. Также м.б. ограничена способность мышц вырабатывать аланин, основную глюконеогенную аминокислоту.
Т.о., в норме у маленьких детей уровень глюкозы в крови падает после 24 ч голодания, концентрация инсулина, соответственно, падает до уровня <5 мкЕд/мл, активируются липолиз и кетогенез, а кетоны могут появляться в моче.
Переход от синтеза гликогена во время и сразу после еды к его распаду и последующему глюконеогенезу регулируется гормонами, при этом центральная роль и значение принадлежат инсулину. После еды концентрация инсулина в плазме повышается до пиковых уровней, в 5-10 раз превышающих их нормальную исходную концентрацию, составляющую -5-10 мкЕд/мл, что снижает концентрацию глюкозы в крови за счет стимуляции синтеза гликогена, усиления поглощения глюкозы периферическими тканями и ингибирования выработки глюкозы. Кроме того, стимулируется липогенез, тогда как липолиз и кетогенез сокращаются. При голодании концентрация инсулина в плазме падает до <5 мкЕд/мл, и вместе с повышением уровня антагонистичных гормонов такое падение инсулина приводит к активации глюконеогенеза (см. рис. выше). Концентрация глюкозы натощак поддерживается за счет активации гликогенолиза и глюконеогенеза, ингибирования синтеза гликогена и активации липолиза и кетогенеза. Следует подчеркнуть, что концентрация инсулина в плазме >5 мкЕд/мл в сочетании с концентрацией глюкозы в крови <40 мг% (2,2 мМЕ) <55 мг/дл (2,8-3,0 мМ) является аномальной, что указывает на состояние чрезмерного действия инсулина, называемое гиперинсулинизмом, вызванным отказом механизмов, которые обычно приводят к подавлению секреции инсулина во время голодания или гипогликемии.
Гипогликемическим эффектам инсулина противодействует ряд гормонов, концентрация которых в плазме увеличивается с падением уровня глюкозы в крови. Эти контррегулирующие гормоны — глюкагон, СТГ, кортизол и адреналин — действуют синергично и согласованно, повышая концентрацию глюкозы в крови за счет активации ферментов гликогенолиза (глюкагон, адреналин); индукции глюконеогенных ферментов (глюкагон, кортизол); подавления поглощения глюкозы мышцами (адреналин, СТГ, кортизол). Кроме того, кортизол усиливает мобилизацию аминокислот из мышц для глюконеогенеза, активируя липолиз и тем самым обеспечивая глицерин для глюконеогенеза и жирные кислоты для кетогенеза (адреналин, кортизол, СТГ, глюкагон). Адреналин также ингибирует секрецию инсулина и стимулирует секрецию СТГ и глюкагона.
Врожденный или приобретенный дефицит любого из данных гормонов встречается редко, но приводит к гипогликемии, которая возникает, когда выработка эндогенной глюкозы не м.б. мобилизована для удовлетворения энергетических потребностей в постабсорбционном состоянии, то есть 4-6 ч у новорожденного и 8-12 ч после еды или во время голодания у младенца или ребенка. Одновременный дефицит нескольких гормонов (гипопитуитаризма с дефицитом АКТГ-кортизола в сочетании с дефицитом СТГ) может привести к гипогликемии, которая считается более серьезной или проявляется ранее во время голодания, нежели чем при изолированном дефиците гормонов. Большинство причин гипогликемии у новорожденных, младенцев и детей связаны с нарушением механизмов адаптации к голоданию в результате: 1) избыточного действия инсулина; 2) неадекватной контррегулирующей гормональной реакцией, в первую очередь кортизола и СТГ; 3) ферментативных нарушений в организме к механизмам хранения и высвобождения гликогена; 4) дефектов глюконеогенеза.
в) Клинические проявления. Клинические признаки гипогликемии обычно делятся на две категории:
1) симптомы, связанные с активацией ВНС и секрецией адреналина, обычно наблюдаемые при быстром снижении концентрации глюкозы в крови, и
2) симптомы, вызванные снижением утилизации глюкозы в ГМ (церебральной гипогликемией), обычно связанным с медленным снижением уровня глюкозы в крови или длительной гипогликемией (табл. 1).
Хотя эти классические симптомы возникают у детей старшего возраста, симптомы гипогликемии у новорожденных и младенцев м.б. более слабыми и проявляться цианозом, апноэ, гипотермией, гипотонией, вялым сосанием, сонливостью и судорогами, что отражает дефицит глюкозы для нормальной деятельности ГМ. Некоторые из этих симптомов м.б. настолько незначительны, что их можно не заметить. Иногда у новорожденных, появившихся естественным путем, гипогликемия может протекать и бессимптомно. Новорожденные с гиперинсулинемией часто являются крупными для гестационного возраста, имитируя черты ребенка, рожденного от матери с плохо контролируемым СД.
Младенцы старшего возраста с гиперинсулинемией могут переедать из-за хронической гипогликемии и становиться тучными. В детстве гипогликемия может проявляться проблемами с пищевым поведением, невнимательностью, повышенным аппетитом или судорогами. Это м.б. ошибочно диагностировано как эпилепсия, опьянение, расстройство личности, головная боль, истерия и задержка в развитии. Содержание глюкозы в крови новорожденных необходимо определять при любых отклонениях от нормы, и если концентрация <55 мг/дл, то сразу же следует проводить коррекцию. В любом возрасте гипогликемию следует рассматривать как причину начального эпизода судорог или внезапного ухудшения психоповеденческих реакций и уровня сознания.
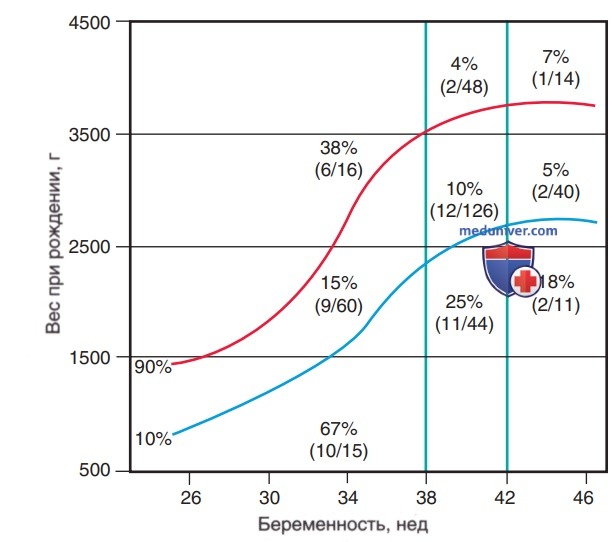
Многие новорожденные страдают бессимптомной (химической) гипогликемией. Частота симптоматической гипогликемии наиболее высока у детей в малом гестационном возрасте (рис. 1). Точную частоту симптоматической гипогликемии установить сложно, поскольку многие симптомы у новорожденных возникают вместе с др. состояниями, такими как инфекции, особенно сепсис и менингит; аномалии ЦНС, кровотечение или отек; гипокальциемия и гипомагниемия; асфиксия; отмена ЛП; апноэ недоношенных; ВПС; полицитемия.
Рисунок 1. Заболеваемость гипогликемией в зависимости от массы тела при рождении, гестационного возраста и в/утробного развития
Начальная симптоматика у новорожденных после рождения варьируется от нескольких часов до недель. В порядке частоты симптомы включают нервозность или тремор, апатию, эпизоды цианоза, судороги, периодические приступы апноэ или тахипноэ, слабый или пронзительный крик, вялость или сонливость, затруднение при приеме пищи (сдавление в груди) и закатывание глаз. Также могут наблюдаться эпизоды потоотделения, внезапной бледности, переохлаждения, остановки сердца и СН. Часто можно отметить скопление эпизодических симптомов. Поскольку эти клинические проявления могут возникать по разным причинам, очень важно измерить уровни глюкозы в сыворотке крови и определить, исчезнут ли симптомы при коррекции уровня глюкозы в крови до нормального уровня; в противном случае следует рассмотреть др. диагнозы.
г) Классификация гипогликемии у младенцев и детей. Классификация основана на знании о регуляции гомеостаза глюкозы у младенцев и детей (табл. 2).
1. Новорожденные, младенцы с гипотрофией и недоношенные младенцы. По оценкам, частота симптоматической гипогликемии среди живорожденных составляет 1-3:1000. Такой рост заболеваемости увеличивается в несколько раз среди новорожденных определенных групп риска (см. табл. 2, рис. 1). Недоношенные дети и младенцы с малым гестационным возрастом предрасположены к развитию гипогликемии.
Факторы, ответственные за высокую частоту гипогликемии в этой группе, а также в др. группах, перечисленных в табл. 2, связаны с недостаточными запасами гликогена в печени, мышечного белка и жира, необходимых для поддержания субстратов, нужных для удовлетворения энергетических потребностей. Такие младенцы маленькие из-за недоношенности или нарушения трансплацентарной передачи питательных в-в. У них недостаточно развита ферментная система глюконеогенеза. Известно, что преходящий гиперинсулинизм, в ответ на диазоксид способствует развитию гипогликемии у детей с асфиксией, малым гестационным возрастом и недоношенных новорожденных. Данная форма гиперинсулинизма, связанная с перинатальной асфиксией, ЗВУР, токсемией у матери и др. перинатальными стрессовыми факторами, вероятно, является наиболее частой причиной гиперинсулинеми-ческой гипогликемии у новорожденных и м.б. довольно тяжелой. У большинства пациентов заболевание проходит быстро, но может сохраняться до 7 мес жизни или дольше.
В отличие от дефицита субстратов или ферментов, гормональная система, по-видимому, нормально функционирует при рождении у большинства новорожденных с низким уровнем риска. Несмотря на гипогликемию, концентрации аланина, лактата и пирувата в плазме выше, что означает снижение скорости их использования в качестве субстратов для глюконеогенеза. Инфузия аланина вызывает дополнительную секрецию глюкагона, но не вызывает значительного повышения уровня глюкозы. В течение первых 24 ч жизни концентрации ацетоацетата и β-гидроксибутирата в плазме у младенцев с малым гестационным возрастом ниже, чем у доношенных, что подразумевает уменьшение запасов липидов, снижение мобилизации жирных кислот, нарушение кетогенеза или сочетание этих состояний. Снижение запасов липидов, скорее всего, происходит из-за того, что кормление новорожденных жиром (триглицеридами) приводит к повышению уровня глюкозы в плазме, кетонов, таких как β-гидроксибутират, и СЖК. Для младенцев с перинатальной асфиксией и новорожденных с малым гестационным возрастом, у которых преходящий гиперинсулинизм, сочетание гипогликемии с пониженными концентрациями β-гидроксибутирата и СЖК является диагностическим признаком гиперинсулинизма.
Важна роль СЖК и их окисления в стимулировании неонатального глюконеогенеза. Поступление СЖК в виде триглицеридов с глюконеогенными предшественниками из смеси или грудного молока при кормлении предотвращает гипогликемию, которая обычно возникает после неонатального голодания. По этим и др. причинам молочное вскармливание вводится рано (при рождении или в течение 2-4 ч) после родов. В условиях больницы, когда кормление невозможно из-за РДС или др. заболевания или когда кормление не поддерживает концентрацию глюкозы в крови на уровне >55 мг/дл целесообразно в/в введение глюкозы со скоростью 4-8 мг/кг в минуту. Младенцы с транзиторной неонатальной гипогликемией обычно могут самопроизвольно поддерживать уровень глюкозы в крови через 2-3 дня жизни, но некоторым требуется более длительная поддержка. У таких поздних младенцев уровни инсулина >5 мкЕд/мл во время гипогликемии следует лечить диазоксидом.
2. Младенцы, рожденные от матерей с сахарным диабетом. Среди транзиторных гиперинсулинемических состояний чаще всего встречаются младенцы, рожденные от матерей с СД. Гестационный СД встречается у ~2% беременных женщин, и 1:1000 беременных страдает инсулинозависимым СД. При рождении младенцы, рожденные от этих матерей, м.б. крупными и полнокровными, а их запасы гликогена, белка и жира в организме переполнены.
Гипогликемия у младенцев от матерей с СД в основном связана с гиперинсулинемией и частично — со снижением секреции глюкагона. Выражена гипертрофия и гиперплазия островков ПЖЖ, а также быстрая двухфазная инсулиновая реакция на глюкозу; такая быстрая инсулиновая реакция отсутствует у здоровых младенцев. Младенцы, рожденные от матерей с СД, имеют недостаточный выброс глюкагона в плазме сразу после рождения, недостаточную секрецию глюкагона в ответ на повышенную симпатическую активность, которая может привести к адреномедуллярному истощению, что отражается в снижении экскреции адреналина с мочой. Вместо нормального гормонального фона плазмы с низким уровнем инсулина, высоким уровнем глюкагона и высоким содержанием катехоламинов отмечаются высокий уровень инсулина, низкий уровень глюкагона и низкий уровень адреналина. Как следствие, выработка эндогенной глюкозы значительно подавляется по сравнению с таковой у здоровых младенцев, что способствует развитию у них гипогликемии.
Матери, у которых мониторинг СД во время беременности, предродовых схваток и родов проводился должным образом, как правило, рожают младенцев, близких к здоровым и с меньшей вероятностью развития неонатальной гипогликемии, а также с др. осложнениями, ранее считавшимися типичными для таких младенцев. При коррекции таких младенцев с гипогликемией экзогенной глюкозой важно избегать гипергликемии, которая вызывает быстрое обильное высвобождение инсулина, что может привести к повторной гипогликемии. Если необходимо, глюкозу следует вводить со скоростью непрерывной инфузии 4-8 мг/кг в минуту, но подходящую дозу для каждого пациента необходимо подбирать индивидуально. Во время родов следует избегать гипергликемии у матери, поскольку она приводит к гипергликемии у плода, провоцирующей гипогликемию, когда при рождении прекращается поступление глюкозы. Гипогликемия, сохраняющаяся на третий день после рождения или первоначально возникающая после 1 нед жизни, требует оценки причин, перечисленных в табл. 2.
Младенцы, рожденные с эритробластозом плода, также могут иметь гиперинсулинемию и много общих физических характеристик, таких как большой размер тела (макросомия), с младенцами, рожденными от матерей с СД. Причина гиперинсулинемии у младенцев с эритробластозом неясна.
3. Постоянная или рецидивирующая гипогликемия у младенцев и детей:
- Гиперинсулинизм. У большинства детей с гиперинсулинизмом, вызывающим гипогликемию, симптомы проявляются в неонатальном периоде или позже в младенчестве. Гиперинсулинизм является наиболее частой причиной стойкой гипогликемии в раннем младенчестве. Младенцы с гиперинсулинизмом обычно крупные при рождении, что связано со стероидными эффектами инсулина во в/утробном периоде. Анамнез или биохимические данные о СД у матери отсутствуют. Симптомы гипогликемии проявляются обычно в первые 18 мес жизни, но иногда у детей старшего возраста.
Концентрация инсулина повышается во время документально подтвержденной гипогликемии; при негиперинсулинемической гипогликемии концентрация инсулина в плазме должна быть <5 мкЕд/мл. У заболевших младенцев концентрация инсулина в плазме во время гипогликемии обычно составляет >5 мкЕд/мл. Некоторые авторы предлагают более жесткие критерии, утверждая, что любое значение инсулина >2 мкЕд/мл при гипогликемии является патологическим. Соотношение «инсулин (мкЕд/мл): глюкоза (мг/дл)» обычно >0,4; уровни белка-1, связывающего инсулиноподобный фактор роста в плазме крови (IGFBP-1), β-гидроксибутирата и СЖК при гиперинсулинизме снижены. Сообщалось о редких случаях активирующих мутаций в сигнальном пути рецептора инсулина, когда клинические и биохимические признаки сходны с состояниями чрезмерной секреции инсулина, но концентрации инсулина низкие до такой степени, что их невозможно обнаружить. Поэтому для описания состояния повышенного действия инсулина предпочтительным термином является гиперинсулинизм. Крупные младенцы могут иметь гипогликемию с первых дней жизни.
У младенцев с меньшей степенью гиперинсулинизма может проявляться гипогликемия только после первых нескольких недель до месяцев, когда частота кормлений уменьшается, особенно по ночам, и гиперинсулинемия препятствует мобилизации эндогенной глюкозы. Наиболее распространенными симптомами являются повышенный аппетит и потребность в кормлении, периоды слабости, нервозность, а также выраженные судороги.
Дополнительные ДК включают быстрое развитие гипогликемии натощак в течение 4-8 ч после голодания, по сравнению с др. причинами гипогликемии (табл. 3, 4); необходимость высокой скорости инфузий экзогенной глюкозы для предотвращения гипогликемии, часто со скоростью >10-15 мг/кг в минуту; отсутствие кетонемии или ацидоза; повышенные уровни С-пептида или проинсулина во время гипогликемии. Последние продукты, связанные с инсулином, отсутствуют при искусственной гипогликемии в результате экзогенного введения инсулина. Гипогликемия неизменно вызывается прекращением кормления на несколько часов, что позволяет одновременно определить содержание глюкозы, инсулина, кетоновых тел и СЖК в одном образце во время клинически проявляющейся гипогликемии. Это называется критической выборкой. Гликемический ответ на введение глюкагона во время гипогликемии показывает резкое увеличение уровня глюкозы не менее чем на 40 мг/дл, что означает сдерживание инсулином мобилизации глюкозы и свидетельствует о сохранности механизмов гликогенолиза (табл. 5-7).
Измерение сывороточной концентрации IGFBP-1 способствует диагностике гиперинсулинемии. Секреция IGFBP-1 резко подавляется действием инсулина; во время гипогликемии, вызванной гиперинсулинизмом, концентрации IGFBP-1 низкие. У пациентов со спонтанной или индуцированной голоданием гипогликемией с низким уровнем инсулина (с кетонемией, нормальный тип голодания) концентрации IGFBP-1 значительно выше.
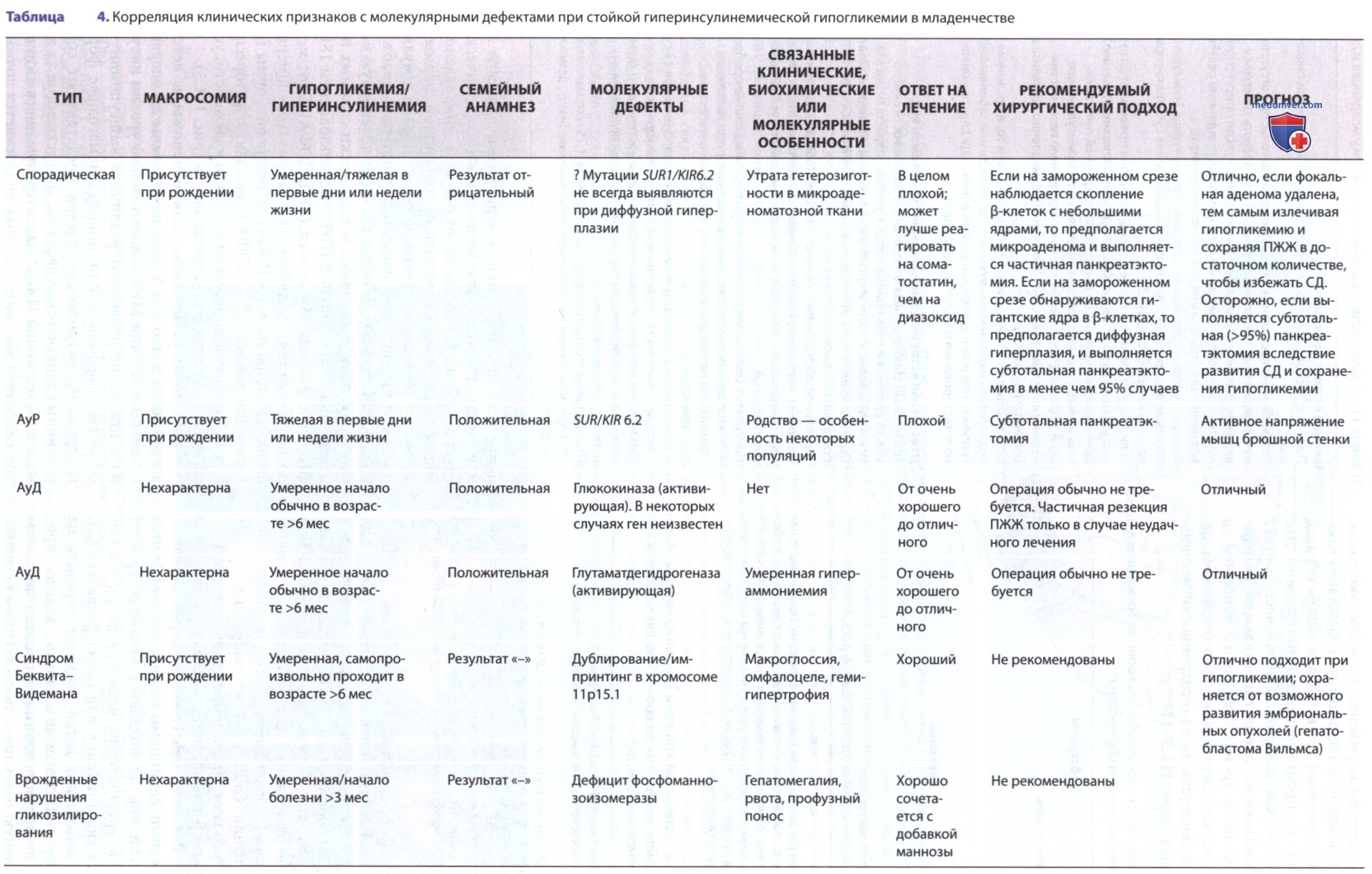
ДД эндогенного гиперинсулинизма включает диффузную гиперплазию β-клеток или очаговую 0-клеточную микроаденому. Различие между этими двумя основными нозологическими единицами важно, т.к. диффузная гиперплазия, если она не поддается лечению, требует почти полной панкреатэктомии несмотря на то, что может сохраняться гипогликемия или позже развиться СД. Некоторые из заболевших детей могут реагировать на сиролимус. Напротив, очаговые аденомы, диагностированные до или во время операции, допускают локализованную радикальную резекцию с последующим нормальным метаболизмом глюкозы. Приблизительно 50% АуР или спорадических форм неонатального/младенческого гиперинсулинизма вызваны очаговыми микроаденомами, которые можно отличить от диффузной формы по характеру реакции инсулина на селективные стимуляторы секреции инсулина, введенные в артерию, снабжающую ПЖЖ, со взятием образцов через печеночную вену. Однако от этих инвазивных и технически сложных процедур отказались в пользу ПЭТ с использованием 18F-L-фтордигидроксифенилаланина (18F-L-ДОФА).
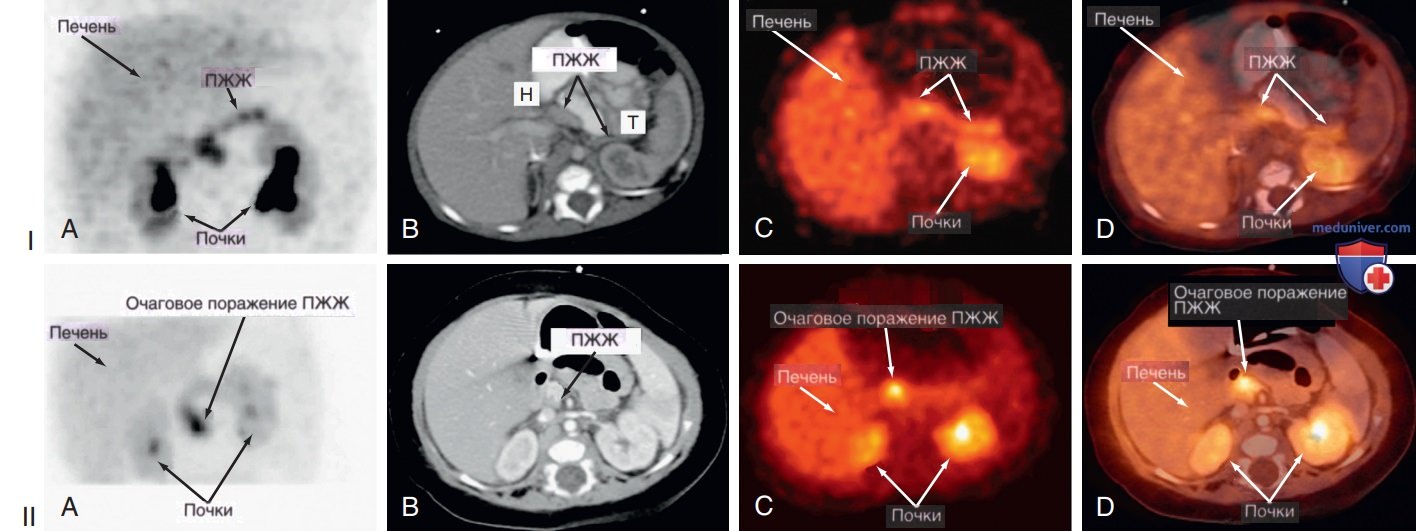
Этот метод позволяет отличить диффузную форму (равномерную флуоресценцию по всей ПЖЖ) от очаговой формы (фокусное поглощение 18F-L-ДОФА и локальную флуоресценцию) с чрезвычайно высокой степенью надежности, успеха, специфичности и чувствительности (рис. 3).
Инсулин-секретирующие макроаденомы в детстве встречаются редко и м.б. диагностированы до операции с помощью КТ или МРТ. Однако только уровни инсулина в плазме не позволяют различить вышеупомянутые объекты. Диффузные или микроаденоматозные формы гиперплазии островковых клеток представляют собой различные генетические дефекты, ответственные за аномалии эндокринной ПЖЖ, характеризующиеся автономной секрецией инсулина, которая не снижается должным образом, когда уровень глюкозы в крови снижается спонтанно или в ответ на провокационные маневры, такие как голодание (см. табл. 4, 7, 8). Клинические, биохимические и молекулярно-генетические подходы позволяют классифицировать врожденный гиперинсулинизм, ранее называвшийся несидиобластозом, на отдельные формы.
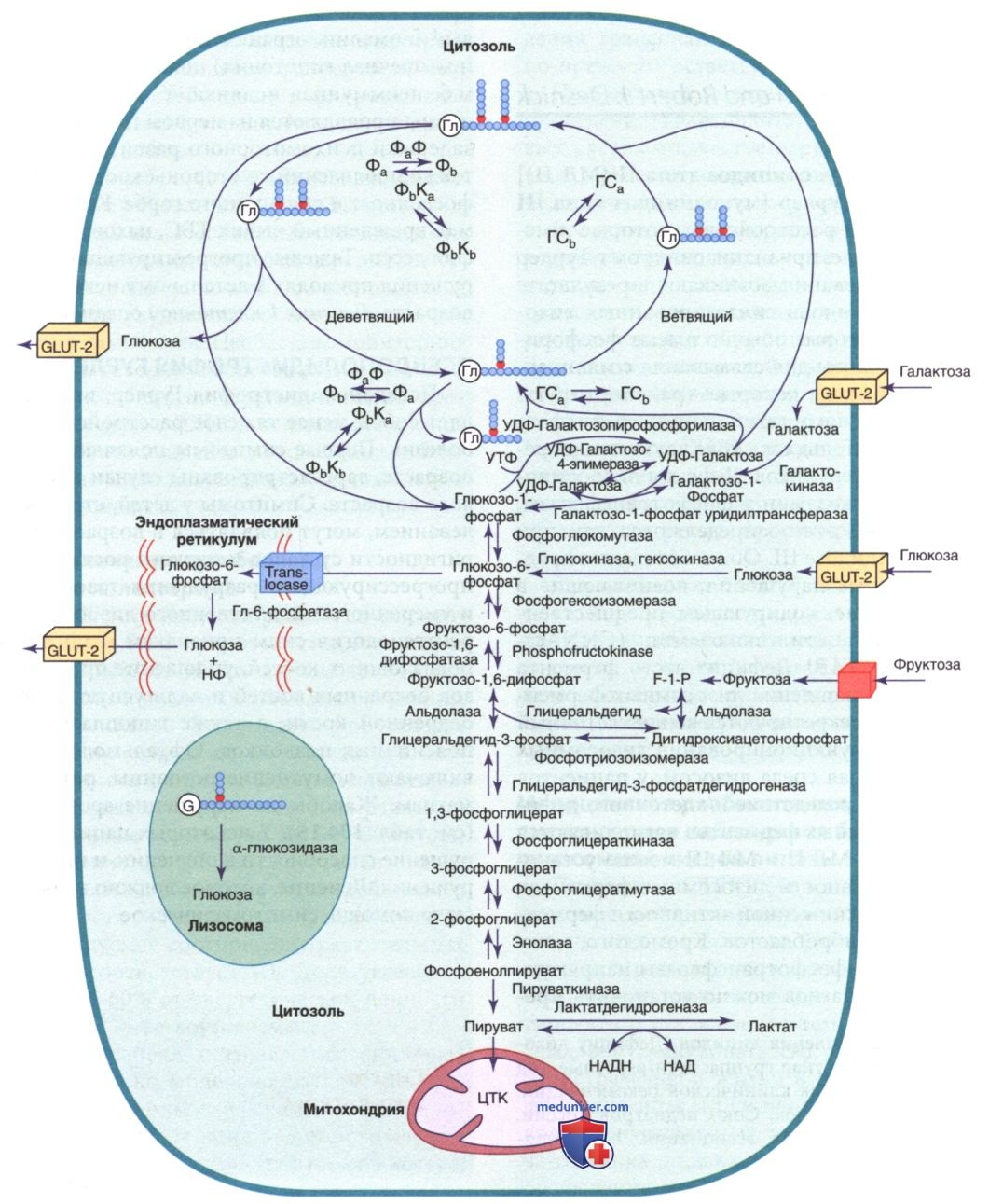
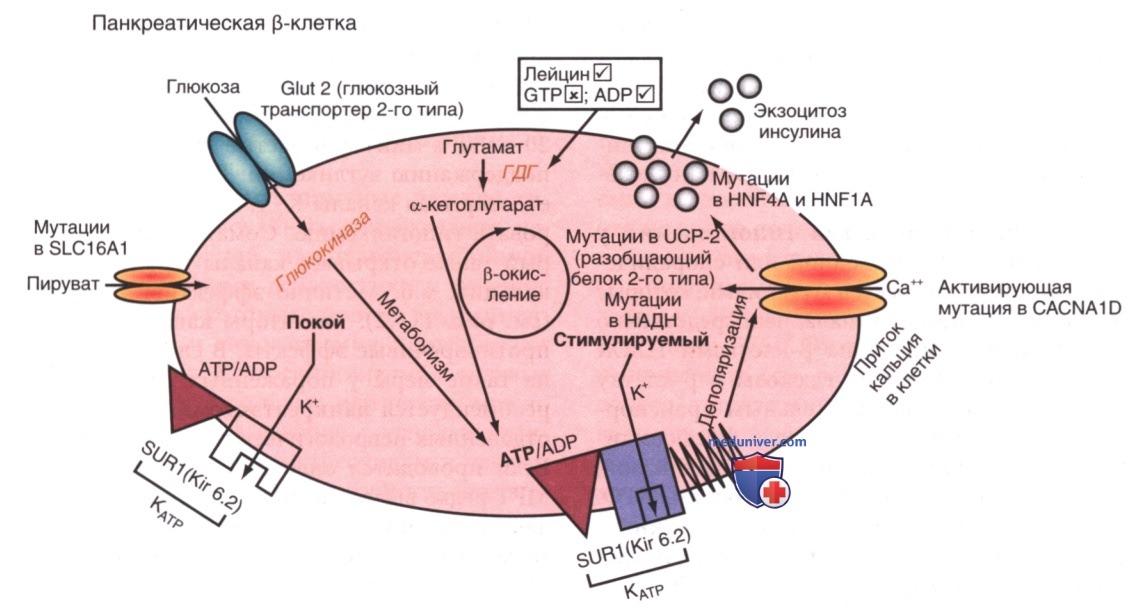
Стойкая гиперинсулинемическая гипогликемия у младенцев (СГГМ) м.б. наследственной или спорадической, является тяжелой и вызывается мутациями, которые влияют на регуляцию калиевого канала, непосредственно участвующего в секреции инсулина β-клетками ПЖЖ (рис. 2). Обычно поступление глюкозы в β-клетку обеспечивается неинсулино-чувствительным транспортером глюкозы GLUT-2. На входе глюкоза фосфорилируется до глюкозо-6-фосфата ферментом глюкокиназой, что позволяет метаболизму глюкозы генерировать АТФ. Повышение молярного отношения АТФ к аденозиндифосфату (АДФ) закрывает АТФ-чувствительный калиевый канал в клеточной мембране (канал КАТФ). Данный канал состоит из двух субъединиц, К-канала внутреннего выпрямления (KIR6.2), входящего в семейство внутренне-ректифицированных калиевых каналов, и регуляторного компонента, тесно связанного с KIR6.2, известного как рецептор сулъфонилмочевины (SUR1). Вместе KIR6.2 и SUR1 составляют калий-чувствительный канал АТФ КАТФ. Обычно КАТФ открыт, но с повышением уровня АТФ и закрытием канала калий накапливается внутри клетки, вызывая деполяризацию мембраны, открытие потенциалзависимых кальциевых каналов, приток кальция в цитоплазму и секрецию инсулина путем экзоцитоза. Гены SUR1 и KIR6.2 расположены близко друг к другу на коротком плече хромосомы 11 — месте гена инсулина.
Рисунок 2. Схема клетки поджелудочной железы (ПЖЖ) с некоторыми важными этапами секреции инсулина. Проникающий через мембрану, чувствительный к АТФ калиевый (К+) канал (КАТФ) состоит из двух субъединиц: рецептора сульфонилмочевины (SUR) и К-канала внутреннего выпрямления (KIR 6.2). В состоянии покоя отношение АТФ к аденозиндифосфату (АДФ) поддерживает КАТФ в открытом состоянии, позволяя отток К+ из клеток. Когда концентрация глюкозы в крови повышается, ее проникновение в β-клетку облегчается транспортером глюкозы Glut-2, но этот процесс не регулируется инсулином. Внутри (3-клетки глюкоза преобразуется в глюкозо-6-фосфат ферментом глюкокиназой, а затем подвергается метаболизму для выработки энергии. В результате возникающее увеличение АТФ по сравнению с АДФ закрывает КАТФ, предотвращая отток К+, а повышение в/клеточного К+ деполяризует клеточную мембрану и открывает кальциевый (Са2+) канал. Повышение в/клеточного Са2+ вызывает секрецию инсулина посредством экзоцитоза. Сульфонилмочевины вызывают секрецию инсулина рецептором (SUR), закрывая КАТФ; диазоксид подавляет этот процесс, тогда как соматостатин или его аналог октреотид подавляют секрецию инсулина, препятствуя притоку кальция в клетки. Генетические мутации в SUR1 или KIR6.2, которые препятствуют открытию КАТФ тонически, поддерживая ненадлежащую секрецию инсулина, ответственны за аутосомно-рецессивные (АуР) формы стойкой гиперинсулинемической гипогликемии младенчества (СГГМ). Одна из аутосомно-доминантных (АуД) форм СГГМ вызвана активирующей мутацией глюкокиназы. Аминокислота лейцин также запускает секрецию инсулина путем закрытия КАТФ. Метаболизму лейцина способствует фермент глутаматдегидрогеназа (ГДГ), а повышенная активность этого фермента в ПЖЖ приводит к гиперинсулинемии с гипогликемией, связанной с гипераммониемией, из-за повышенной активности ГДГ в печени. Мутации в пируватном канале SLC16A1 могут вызывать эктопическую экспрессию в β-клетках и позволяют пирувату, накопленному во время ФН, вызывать секрецию инсулина и, т.о., гипогликемию, обусловленную ФН. Мутации митохондриального разобщающего белка 2 (UCP2) и HADH связаны с гиперинсулинизмом по механизмам, которые еще предстоит определить. Мутации в факторах транскрипции ядерных факторов гепатоцитов 4α и 1α м.б. связаны с макросомией новорожденных и гиперинсулинизмом, но в более позднем возрасте прогрессируют до моногенной формы СД у подростков. Активирующие мутации в кальциевом канале CACNA1D допускают приток кальция в клетки и, следовательно, нерегулируемую секрецию инсулина при мембранных напряжениях, которые обычно исключают приток кальция. √ — стимуляция; ГТФ — гуанозинтрифосфат; X — торможение
Рисунок 3. Врожденный гиперинсулинизм. I — панели (диффузные): позитронно-эмиссионная томография (ПЭТ) с фтором 18 (18F)-L-ДОФА у пациента с диффузной формой врожденного гиперинсулинизма: A — диффузное поглощение 18F-L-ДОФА визуализируется по всей поджелудочной железе (ПЖЖ). Поперечные сечения показывают B — нормальную ткань ПЖЖ на компьютерной томографии (КТ) органов брюшной полости (ОБП); C — диффузное поглощение 18F-L-ДОФА ПЖЖ; D — подтверждение поглощения 18F-L-ДОФА панкреатитом при совместной регистрации; Н — головка ПЖЖ; Т — хвост ПЖЖ. II — панели (очаговые): ПЭТ 18F-L-ДОФА у пациента с очаговой формой врожденного гиперинсулинизма; A — дискретная зона повышенного поглощения 18F-L-ДОФА визуализируется в головке ПЖЖ. Интенсивность этой области больше, чем наблюдается в печени и соседней нормальной ткани ПЖЖ. Поперечные сечения показывают B — нормальную ткань ПЖЖ на КТ ОБП; C — очаговое поглощение 18F-L-ДОФА головкой ПЖЖ; D — подтверждение поглощения 18F-L-ДОФА головкой ПЖЖ с совместной регистрацией
Инактивирующие мутации в гене SUR1 или, менее частые, KIR6.2 препятствуют открытию калиевого канала; он остается закрытым с постоянной деполяризацией и, следовательно, постоянным потоком кальция внутри. Т.о., секреция инсулина является непрерывной и не зависит от концентрации глюкозы. Сообщается также о более легкой АуД-форме этих дефектов. Точно так же активирующая мутация в гене активности фермента глюкокиназы или глутаматдегидрогеназы увеличивает метаболизм субстрата и приводит к закрытию калиевого канала из-за избыточного производства АТФ, что вызывает гиперинсулинизм. Генетические дефекты метаболизма жирных кислот, факторов транскрипции инсулина — гепатоцитарного ядерного фактора 4-альфа (HNF-4α; англ. Hepatocyte nuclear factor 4 alpha), 1-альфа (HNF-1α) и разобщающего белка (UCP-2; англ. An uncoupling protein) — комплекса митохондриальных генов, также участвуют в гиперинсулинемической гипогликемии. Совсем недавно сообщалось об активирующей мутации в кальциевом канале, которая обеспечивает приток кальция в β-клетку, что приводит к чрезмерной, нерегулируемой секреции инсулина и гипогликемии, реагирующей на диазоксид. Инактивирующие мутации гена глюкокиназы или активирующие мутации КАТФ, которые предотвращают или ограничивают закрытие канала, ответственны за неадекватную секрецию инсулина и составляют основу некоторых форм СД зрелого возраста у молодых и неонатального СД.
Наследственные формы СГГМ более распространены в определенных популяциях, особенно в арабских и еврейских общинах ашкенази, где заболеваемость может достигать 1:2500, по сравнению со спорадическими показателями в общей популяции 1:50 000. Данные АуР-формы СГГМ обычно проявляются в ближайшем послеродовом периоде у крупного плода с МТ >4,0 кг и тяжелой рецидивирующей или стойкой гипогликемией, проявляющейся в первые часы или дни жизни. Инфузии глюкозы до 15-20 мг/кг в минуту и частые кормления не способствуют поддержанию эугликемии. Диазоксид, который действует, открывая каналы КАТф, не может адекватно контролировать гипогликемию. Соматостатин (октреотид), который также открывает каналы КАТФ и ингибирует приток кальция, м.б. частично эффективным у 50% пациентов (см. рис. 2). Блокаторы кальциевых каналов имеют противоречивые эффекты. В случае отсутствия реакции на такие меры у пораженных пациентов настоятельно рекомендуется панкреатэктомия с целью профилактики отдаленных неврологических последствий гипогликемии.
Если проводится операция, предоперационные КТ или МРТ редко выявляют изолированную аденому, что позволяет локально ее резецировать. Интраоперационное УЗИ может выявить небольшую непальпируемую аденому, что позволяет провести частичную резекцию. Аденомы часто появляются в младенчестве или раннем детстве.
Отличить очаговые и диффузные случаи стойкого гиперинсулинизма пытались несколькими способами. Перед операцией чреспеченочная катетеризация воротной вены и выборочный забор крови из панкреатической вены для измерения инсулина могут локализовать очаговое поражение в результате повышения концентрации инсулина в определенном месте. Селективная катетеризация артериальных ветвей, снабжающих ПЖЖ, с последующей инфузией стимуляторов секреции, таких как кальций, и забор образцов из воротной вены для измерения концентрации инсулина (стимуляция артерий — забор венозных образцов) могут локализовать поражение. Оба подхода являются высокоинвазивными, ограниченными специализированными центрами и не всегда успешными в определении отличий очаговых форм от диффузных. Т.о., эти методы не рекомендуются, и в большей степени от них отказались. L-ДОФА, меченная фтором 18 (18F), в сочетании с ПЭТ-сканированием является очень многообещающим способом отличить очаговые поражения от диффузных при гиперинсулинизме, не поддающиеся лечению (см. рис. 3). Золотым стандартом остается интраоперационное гист. исследование. Диффузный гиперинсулинизм характеризуется наличием крупных β-клеток с аномально большими ядрами, тогда как очаговые аденоматозные поражения демонстрируют малые и нормальные β-клеточные ядра.
Хотя мутации SUR1 присутствуют в обоих типах, очаговые поражения возникают в результате случайной потери материнского импринтированного роста ингибирующего гена на материнской хромосоме 11р в сочетании с отцовской передачей мутировавшей отцовской хромосомы SUR1 или KIR6.2 11р, экспрессирующей инсулин, как ген фактора роста-2 (IGF2). Т.о., очаговая форма представляет собой двойной удар — потерю материнского репрессора и передачу отцовской мутации, содержащей ген, способствующий росту. Это аналогично тому, что происходит у детей с гиперинсулинемической гипогликемией, наблюдаемой при синдроме Беквита-Видемана, как обсуждается ниже.
Локальное удаление очаговой гиперплазии аденоматозных островковых клеток приводит к излечению с очень редкими рецидивами. При диффузной форме рекомендуется субтотальная резекция (85-90% объема) ПЖЖ. Однако почти тотальная панкреатэктомия, необходимая при диффузных гиперпластических поражениях, часто связана со стойкой гипогликемией или более поздним развитием гипергликемии, требующей инсулинотерапии.
Дальнейшая резекция оставшейся ПЖЖ может потребоваться, если гипогликемия сохраняется и не устраняется такими медикаментозными средствами, как применение октреотида или диазоксида.
Операцию должны проводить опытные детские хирурги в медцентрах, оборудованных для оказания необходимой предоперационной и послеоперационной помощи всеми возможностями диагностики и лечения. У некоторых пациентов, которых лечили медикаментозно, гиперинсулинизм и гипогликемия регрессируют в течение многих месяцев. Если гипогликемия впервые проявляется в возрасте от 3 до 6 мес или позже, можно попытаться провести пробное лечение с использованием мед. подходов с диазоксидом, октреотидом и частым кормлением в течение 2-4 нед. Неспособность поддержать эугликемию и появление нежелательных побочных эффектов от ЛС могут вызвать необходимость хирургического вмешательства. Сообщалось о некоторых успехах в подавлении высвобождения инсулина и коррекции гипогликемии у пациентов с СГГМ при использовании октреотида длительно действующего аналога соматостатина. Большинство случаев неонатальной СГГМ носят спорадический характер; наследственные формы позволяют проводить генетическое консультирование на основе ожидаемого АуР-наследования.
Вторая форма наследственной СГГМ предполагает АуД-наследование. Клинические признаки, как правило, менее серьезны, и более вероятно развитие гипогликемии в ближайшем послеродовом периоде обычно после периода отлучения от груди, в среднем в возрасте до 1 года. При рождении макросомия наблюдается редко, а реакция на диазоксид почти одинакова. Первоначальное проявление болезни м.б. отсроченным и в редких случаях может проявиться даже в 30-летнем возрасте, за исключением провоцирования голоданием. Генетическая основа этой АуД-формы не определена. Она не всегда связана с мутациями генов KIR6.2/SUR1. Активирующая мутация гена глюкокиназы передается по АуД-типу. При отягощенной наследственности м.б. проведено генетическое консультирование, исходящее из 50% вероятности случаев рецидива у будущего потомства.
Третья форма стойкой СГГМ сопровождается легкой и бессимптомной гипераммониемией, как правило, в отдельных случаях, хотя бывает и АуД-наследование. Проявление больше похоже на АуД-форму, чем на АуР. Диета и диазоксид устраняют симптомы, но некоторым пациентам может потребоваться панкреатэктомия. Связь гиперинсулинизма и гипераммониемии вызвана унаследованной или мутацией de novo, обусловленной увеличением функции фермента глутаматдегидрогеназы. Результирующее усиление окисления глутамата в β-клетках ПЖЖ увеличивает концентрацию АТФ и, следовательно, соотношение АТФ/АДФ, которое закрывает КАТф, что приводит к деполяризации мембраны, притоку кальция в клетки и усилению секреции инсулина (см. рис. 2). В печени чрезмерное окисление глутамата до β-кетоглутарата может приводить к образованию аммиака и препятствует превращению глутамата в N-ацетилглутамат, который является важным кофактором для удаления аммиака через цикл мочевины путем активации фермента карбамоилфосфатсинтетазы. Легкая гипераммониемия при концентрации 100-200 мкМ/л не вызывает симптомов или последствий для ЦНС, характерных для др. состояний гипераммониемии. Лейцин, мощная аминокислота для стимуляции секреции инсулина и участвующая в лейцин-чувствительной гипогликемии, действует путем аллостерической стимуляции глутаматдегидрогеназы.
Т.о., лейцин-чувствительная гипогликемия, м.б. формой синдрома гиперинсулинемии-гипераммониемии и способствовать легким нарушениям канала КАТФ, что не всегда должно быть связано с умеренным увеличением аммиака в сыворотке крови.
Гипогликемия, связанная с гиперинсулинемией, наблюдается у 50% пациентов с синдромом Беквита-Видемана. Этот синдром вызван расстройством импринтинга и характеризуется омфалоцеле, гигантизмом, макроглоссией, микроцефалией и висцеромегалией (рис. 4). Отмечаются характерные боковые трещины мочки уха и «пылающий» невус на лице; а также у многих из этих младенцев — гемигипертрофия. Диффузная гиперплазия островковых клеток ПЖЖ встречается у младенцев с гипогликемией. Диагностические и терапевтические подходы такие же, как обсуждавшиеся ранее, хотя микроцефалия и задержка развития ГМ могут возникать независимо от гипогликемии. Пациенты с синдромом Беквита-Видемана предрасположены к опухоли, включая опухоль Вильмса, гепатобластому, карциному надпочечников, гонадобластому и рабдомиосаркому. Этот синдром чрезмерного роста вызван мутациями в области хромосомы 11р15.5, близкой к генам инсулина SUR1, KIR6.2 и IGF2. Дупликации в этой области и генетический импринтинг дефектной или отсутствующей копии материнского гена вовлечены в вариабельные особенности и паттерны передачи. Гипогликемия может исчезнуть за несколько недель или месяцев лечения, а резекция ПЖЖ может потребоваться крайне редко.
Рисунок 4. Синдром Беквита-Видемана
Синдром Кабуки, вызванный мутациями метилтрансферазы или деметилазы, является второй по распространенности синдромной формой гиперинсулинемической гипогликемии младенчества после синдрома Беквита-Видемана. Неонатальная гипогликемия с врожденным гиперинсулинизмом встречается у ~70% детей с этим синдромом; большинство из них реагируют на диазоксид. Сообщается, что врожденная гиперинсулинемия также возникает при синдроме Тернера. Сообщалось, что активирующие мутации в АКТ2 и PI3-киназе инсулинового сигнального каскада связаны с гипокетотической гипогликемией и др. метаболическими особенностями, указывающими на чрезмерное действие инсулина, но концентрации инсулина опускается ниже нормы в результате отрицательной обратной связи от сигнала активированного рецептора инсулина.
Сообщается, что гиперинсулинемическая гипогликемия младенчества является проявлением одной формы врожденного нарушения гликозилирования. Нарушения гликозилирования белков обычно проявляются неврологическими симптомами, но могут также включать дисфункцию печени с гепатомегалией, трудноизлечимую диарею, энтеропатию с потерей белка и гипогликемию. Такие расстройства часто не диагностируются. Единственное заболевание, связанное с гиперинсулинемической гипогликемией младенчества, вызванное дефицитом изомеразы фосфоманнозы. Для клинического улучшения достаточно дополнительного лечения пероральной маннозой, 0,17 г/кг 6 р/сут.
После первых 12 мес жизни гиперинсулинемические состояния возникают редко, пока как следствие не появятся аденомы островковых клеток, когда пациенту исполнится несколько лет. Гиперинсулинемию в результате аденомы островковых клеток следует рассматривать у любого ребенка >5 лет, у которого имеется гипогликемия. Аденомы островковых клеток не «контрастируются» во время сканирования L-ДОФА, меченная фтором-18 (18F). Аденома островковых клеток у ребенка должна вызывать подозрение на возможность множественной эндокринной неоплазии типа I (синдром Вермера), которая включает мутации в гене менина и м.б. связана с гиперпаратиреозом и опухолями гипофиза. В табл. 7 и 8 описан диагностический подход. У новорожденного голодание в течение всего 6-8 ч (один пропущенный прием пищи в 3-4-часовом графике кормления) м.б. достаточным, чтобы спровоцировать гипогликемию, и этот маневр следует выполнять для исключения стойких форм гипогликемии, перед выпиской из неонатологического отделения.
У младенцев и детей старшего возраста голодание продолжительностью до 24-36 ч обычно вызывает гипогликемию; сопутствующая гиперинсулинемия подтверждает диагноз при условии, что искусственное введение инсулина родителями исключено. Иногда могут потребоваться провокационные тесты. Экзогенно вводимый инсулин можно отличить от эндогенного инсулина путем одновременного измерения концентрации С-пептида. Если уровни С-пептида повышены, за гипогликемию ответственна секреция эндогенного инсулина; если уровни С-пептида низкие, а уровни инсулина высокие, был введен экзогенный инсулин, возможно, как форма жестокого обращения с детьми. Аденомы островковых клеток в этом возрасте лечат хирургическим путем. АТл к инсулину или рецепторам инсулина (инсулиноподобное действие) редко связаны с гипогликемией. Некоторые опухоли секретируют инсулиноподобный фактор роста, взаимодействующий с рецепторами инсулина, тем самым вызывая гипогликемию. Проницательный врач должен учитывать возможность преднамеренного или случайного проглатывания ЛС, таких как сульфонилмочевина или родственное соединение, которые стимулируют секрецию инсулина. В таких случаях концентрация как инсулина, так и С-пептида в крови будет повышена. При приеме ЛС следует учитывать случайную замену ЛС, усиливающего секрецию инсулина, и ошибки в дозировке, от которых внезапно развивается задокументированная гипогликемия.
Сообщалось о редкой форме гиперинсулинемической гипогликемии после ФН. В то время как глюкоза и инсулин остаются неизменными, после умеренных непродолжительных упражнений у некоторых пациентов развивается тяжелая гипогликемия с гиперинсулинемией через 15-50 мин после тех же стандартных упражнений. Эта форма гиперинсулинизма, вызванного ФН, возникает из-за аномальной реакции высвобождения инсулина β-клетками на пируват, образующийся во время физических упражнений. Ген, ответственный за этот синдром, SLC16A1, регулирует монокарбоксилат транспортер-1 (MCT1R; англ. Monocarboxylate transporter 1), который контролирует проникновение пирувата в клетки. Доминантные мутации в SLC16A1, которые увеличивают эктопическую экспрессию MCTR1 в [3-клетках ПЖЖ, допускают избыточное проникновение пирувата в β-клетки и действуют, увеличивая секрецию инсулина, что приводит к гипогликемии во время физических упражнений.
После бариатрических операций по поводу ожирения редко наблюдалась гипогликемия с так называемым неси-диобластозом. Механизм этой формы гиперинсулинемической гипогликемии еще предстоит определить.
Младенцы и дети с фундопликацией Ниссена — относительно распространенной процедурой, используемой для улучшения гастроэзофагеального рефлюкса, — часто имеют связанный с гипогликемией демпинг-синдром. Характерные признаки включают значительную гипергликемию 200 мг/дл и до 500 мг/дл через 30 мин после приема пищи и тяжелую гипогликемию (в среднем 32 мг/дл в одной серии) через 1,5-3,0 ч. Ранняя фаза гипергликемии связана с быстрым и избыточным высвобождением инсулина, что вызывает возвратную гипогликемию. Была предложена роль повышенной секреции глюканоподобного пептида-1 (GLP1; англ. Glucagon-like peptide 1), и сообщалось, что в некоторых случаях уровни глюкагона были крайне низкими. Однако не всегда ясно понимаются физиологические механизмы, и попытки лечения не всегда эффективны. Как сообщалось, использование акарбозы, ингибитора абсорбции глюкозы, было успешным в одной небольшой серии.
4. Эндокринная недостаточность. Гипогликемия, связанная с эндокринной недостаточностью, обычно вызвана надпочечниковой недостаточностью с дефицитом СТГ или без него. При пангипопитуитаризме, изолированном АКТГ, или дефиците СТГ, или комбинированном дефиците АКТГ и СТГ, частота гипогликемии достигает 20%. У новорожденных гипогликемия м.б. признаком гипопитуитаризма. У мальчиков микрофаллос может указывать на сопутствующий дефицит гонадотропина. Новорожденные с гипопитуитаризмом часто страдают формой гепатита, связанной с холестатической желтухой и гипогликемией. Комбинация гипогликемии и холестатической желтухи требует исключения гипопитуитаризма как причины их возникновения, т.к. желтуха проходит при заместительной терапии СТГ, кортизолом и при необходимости тиреоидином. Такая констелляция часто ассоциируется с синдромом септооптической дисплазии.
Когда заболевание надпочечников тяжелое, напр. при врожденной гиперплазии надпочечников, вызванной нарушениями функции ферментов синтеза кортизола, при кровоизлиянии в надпочечники или врожденной гипоплазии надпочечников диагностическими признаками служат электролитные нарушения в сыворотке крови с гипонатриемией и гиперкалиемией или нарушения развития половых органов, (см. главу 576). У детей старшего возраста задержка роста должна указывать на дефицит СТГ. Гиперпигментация, слабость или тяга к соли могут указывать на первичную недостаточность надпочечников (болезнь Аддисона), характеризующуюся значительным повышением уровня АКТГ или невосприимчивостью надпочечников к экзогенному АКТГ, вызванной дефектом рецептора надпочечников для АКТГ, врожденной гипоплазией надпочечников, адренолейкодистрофией или синдромом Оллгрова (синдром трех А). Следует учитывать частую связь болезни Аддисона в детстве с гипопаратиреозом (гипокальциемией), хроническим кожно-слизистым кандидозом и др. эндокринопатиями, которые составляют синдром аутоиммунной полиэндокринопатии 1-го типа. Адренолейкодистрофия и врожденная гипоплазия надпочечников связаны с полом и должны учитываться при ДД первичной болезни Аддисона у детей мужского пола (см. раздел 104.2).
Гипогликемия при дефиците кортизола-СТГ м.б. вызвана снижением уровня глюконеогенных ферментов при дефиците кортизола, повышенным использованием глюкозы, отсутствием антагонистических эффектов СТГ на действие инсулина или неспособностью обеспечить эндогенный глюконеогенный субстрат в форме аланина и лактата с компенсирующим расщеплением жира и образованием кетонов. Дефицит этих гормонов приводит к снижению уровня глюконеогенного субстрата, что напоминает синдром кетотической гипогликемии. Следовательно, обследование ребенка с гипогликемией требует исключения дефицита АКТГ-кортизола или СТГ и в случае диагностирования — его соответствующей замены кортизолом или СТГ.
Теоретически причиной гипогликемии м.б. дефицит адреналина. Выведение адреналина с мочой было снижено у некоторых пациентов со спонтанной или инсулиноиндуцированной гипогликемией, у которых также отмечалось отсутствие бледности кожных покровов и тахикардии. Это говорит о том, что нарушение высвобождения катехоламинов в результате дефекта в любом месте вдоль гипоталамо-вегетативно-адреномедуллярной оси м.б. причиной гипогликемии. Однако эта возможность была оспорена из-за редкости гипогликемии у пациентов с двусторонней адреналэктомией при условии, что они получают адекватную замену ГКС, а также из-за того, что сниженная экскреция адреналина обнаруживается у здоровых пациентов с повторной инсулиноиндуцированной гипогликемией. Многие пациенты, у которых описана гипогликемия с недостаточностью выведения адреналина, соответствуют критериям кетотической гипогликемии (см. далее). Кроме того, повторяющаяся гипогликемия приводит к снижению реакций кортизола и адреналина, что чаще всего наблюдается при инсулинозависимом СД и синдроме нечувствительности к развитию гипогликемии, связанном с вегетативной недостаточностью.
Теоретически дефицит глюкагона у младенцев или детей м.б. связан с гипогликемией, но такие случаи нигде не были зарегистрированы.
5. Этиологии, связанные с ограничением субстрата:
- Кетотическая гипогликемия. Идиопатическая кетотическая гипогликемия является наиболее частой формой детской гипогликемии. Такое состояние обычно проявляется в возрасте от 18 мес до 5 лет и проходит спонтанно к 8-9 годам. Эпизоды гипогликемии обычно возникают в периоды интеркуррентного заболевания, когда прием пищи ограничен. Классический анамнез: ребенок, который плохо ест или полностью избегает ужина, его трудно разбудить на следующее утро, и, т.о., он снова плохо ест, к середине утра у него м.б. припадок или коматозное состояние. Др. распространенное проявление болезни возникает, когда родители поздно ложатся спать, а больной ребенок не может позавтракать, что продлевает ночное голодание.
Во время документально подтвержденной гипогликемии наблюдается выраженная кетонурия и кетонемия. Концентрации инсулина в плазме соответственно низкие, <5 мкЕд/мл, что исключает гиперинсулинемию. Кетогенно-провокационная диета, ранее являвшаяся диагностическим тестом, больше не используется для установления диагноза, потому что голодание у восприимчивых людей само по себе провоцирует эпизод гипогликемии с кетонемией и кетонурией в течение 12-18 ч. Нормальные дети того же возраста могут выдерживать голодание без развития гипогликемии в течение того же периода, хотя даже они могут иметь такие признаки к 36 ч голодания.
У детей с кетотической гипогликемией концентрации аланина в плазме крови заметно снижаются в базальном состоянии после ночного голодания и еще больше — при длительном голодании. Аланин, вырабатываемый в мышцах, является основным предшественником глюконеогенных в-в. Аланин — единственная аминокислота, содержание которой значительно ниже у этих детей, а инфузии аланина (250 мг/кг) быстро повышают уровень глюкозы в плазме, не вызывая значительных изменений уровней лактата или пирувата в крови, что указывает на то, что весь глюконеогенный путь не зависит от уровня пирувата, но отмечается дефицит субстрата. Гликогенолитические пути также не нарушены, поскольку глюкагон вызывает нормальный гликемический ответ у заболевших детей в сытом состоянии. Уровни гормонов, противодействующих гипогликемии, соответственно повышены, а уровень инсулина — соответственно низкий.
Этиология кетотической гипогликемии м.б. дефектом на любом из сложных этапов катаболизма белка, окислительного дезаминирования аминокислот, трансаминирования, синтеза аланина или оттока аланина из мышц. Дети с кетотической гипогликемией часто меньше, чем дети контрольной группы того же возраста, имеют в анамнезе преходящую неонатальную гипогликемию. Любое снижение мышечной массы может существенно снизить поставку глюконеогенного субстрата, в то время когда потребность в глюкозе на единицу МТ уже относительно высока, что предрасполагает пациента к быстрому развитию гипогликемии, при этом кетоз представляет собой попытку переключиться на поставку альтернативной энергии. Дети с кетотической гипогликемией плохо переносят голодание. Аналогичная относительная непереносимость голодания присутствует у нормальных детей, которые не могут поддерживать уровень глюкозы в крови после 30-36 ч голодания, по сравнению со способностью взрослых к длительному голоданию. Хотя дефект может присутствовать при рождении, он может не проявиться до тех пор, пока ребенок не будет подвержен стрессу вследствие более длительных периодов ограничения поступления энергии. Более того, спонтанная ремиссия, наблюдаемая у детей в возрасте 8-9 лет, м.б. объяснена увеличением мышечной массы с последующим увеличением запаса эндогенного субстрата и, с возрастом, относительным снижением потребности в глюкозе на единицу МТ.
В ожидании спонтанного разрешения этого синдрома лечение кетотической гипогликемии состоит из частых приемов пищи с высоким содержанием белка и углеводов. При интеркуррентных заболеваниях родителей следует научить проверять мочу ребенка на наличие кетоновых тел, появление которых на несколько часов опережает гипогликемию. При кетонурии ребенку следует предложить сладкое питье с высоким содержанием углеводов. При непереносимости ребенка следует госпитализировать и лечить в/в введением глюкозы.
- Разветвленно-цепочечная кетонурия (болезнь «кленового сиропа»). См. отдельную статью на сайте - просим Вас пользоваться формой поиска по сайту выше.
Когда-то эпизоды гипогликемии приписывались высокому уровню лейцина, но данные свидетельствуют о том, что нарушение выработки аланина и его доступность в качестве глюконеогенного субстрата во время голодания ответственны за гипогликемию.
6. Гликогеноз. См. отдельную статью на сайте - просим Вас пользоваться формой поиска по сайту выше.
- Дефицит глюкозо-6-фосфатазы (гликогеновой болезни I типа). Больные дети обычно хорошо переносят хроническую гипогликемию. Классические симптомы гипогликемии отсутствуют даже при уровне глюкозы в крови в диапазоне 20-50 мг/дл, что отражает адаптацию ЦНС к кетоновым телам и лактату в качестве альтернативной энергии. Гепатомегалия и слабый рост являются постоянными физическими особенностями. Гипогликемия связана с ацидозом (НСО3 <18 мэкв/л) и повышенным содержанием β-гидроксибутирата и лактата; часто наблюдается гиперурикемия. Управление подробно обсуждается в отдельной статье на сайте - просим Вас пользоваться формой поиска по сайту выше.
- Дефицит амило-1,6-глюкозидазы (дефицит гликоген-деветвящего фермента; гликогеновой болезни III типа). См. отдельную статью на сайте - просим Вас пользоваться формой поиска по сайту выше.
- Дефицит фосфорилазы печени (гликогеновой болезни VI типа). Низкая активность фосфорилазы печени м.б. результатом дефекта на любом из этапов активации; было описано множество дефектов. Наблюдаются гепатомегалия, чрезмерное накопление гликогена в печени, задержка роста и эпизодическая симптоматическая гипогликемия. Диета с высоким содержанием белка и низким содержанием углеводов обычно предотвращает гипогликемию.
- Дефицит гликоген-синтетазы. Нарушения синтеза гликогена встречается крайне редко. Гипогликемия и гиперкетонемия отмечаются натощак, поскольку запасы гликогена сильно уменьшены или отсутствуют. Однако после еды возникает гипергликемия с глюкозурией из-за неспособности ассимилировать часть глюкозы в гликоген. Во время гипогликемии натощак уровни контррегулирующих гормонов, включая катехоламины, соответственно повышены или нормальны, а уровни инсулина соответственно понижены. Печень не увеличена. Диета, богатая белком, и частый прием пищи приводят к значительному клиническому улучшению, включая скорость роста. Дефицит гликогенсинтетазы имитирует синдром кетотической гипогликемии и должен учитываться при ДД этого синдрома.
7. Нарушения гликонеогенеза:
- Недостаточность фруктозо-1,6-дифосфатазы. См. отдельную статью на сайте - просим Вас пользоваться формой поиска по сайту выше.
Дефицит данного фермента приводит к блокированию глюконеогенеза от всех возможных предшественников на уровне превращения фруктозо-1,6-дифосфата. Инфузия данных глюконеогенных предшественников приводит к лактат-ацидозу без повышения уровня глюкозы. Угнетение гликогенолиза провоцирует острый приступ гипогликемии. Гликогенолиз остается неизменным, а глюкагон вызывает нормальный гликемический ответ в сытом состоянии, но не натощак. Поэтому у больных гипогликемия возникает только во время дефицита калорий, напр. при голодании или во время интеркуррентных заболеваний. Пока запасы гликогена остаются в норме, гипогликемия не развивается. В семейном анамнезе могут отмечаться случаи гибели грудных детей с установленной гепатомегалией от необъяснимого метаболического ацидоза.
- Дефекты окисления жирных кислот. См. отдельную статью на сайте - просим Вас пользоваться формой поиска по сайту выше. Важная роль окисления жирных кислот в поддержании глюконеогенеза подчеркивается примерами врожденных или вызванных лекарствами дефектов метаболизма жирных кислот, которые м.б. связаны с гипогликемией при голодании.
Разл. врожденные дефициты ферментов вызывают нарушение метаболизма карнитина или жирных кислот. Тяжелая и относительно распространенная форма гипогликемии голодания с гепатомегалией, кардиомиопатией и гипотонией возникает при дефиците КоА-дегидрогеназы длинно- и среднецепочечных жирных кислот. Уровни карнитина в плазме низкие, кетоны отсутствуют, но в моче присутствует дикарбоновая ацидурия. Клинические проявления у пациентов с дефицитом ацил-КоА-дегидрогеназы сходны с синдром Рейе. Отмечаются рецидивирующие эпизоды тяжелой гипогликемической комы при голодании и остановке кровообращения (явления, подобные синдрому внезапной детской смерти). Также у пациентов с множественными нарушениями ацил-КоА-дегидрогеназы встречаются тяжелая гипогликемия и метаболический ацидоз без кетонемии. Др. клиническими признаками являются гипотония, судороги и резкий запах тела.
Выживаемость зависит от того, являются ли дефекты серьезными или легкими; диагноз устанавливается на основании исследований активности ферментов в биопсии ткани печени или культивированных фибробластах заболевших пациентов.
Тандемная масс-спектрометрия может использоваться для проб крови, даже проб на фильтровальной бумаге, для скрининга врожденных пороков. Также молекулярная диагностика доступна для большинства субъектов. Частота дефицита ацил-КоА-дегидрогеназы составляет > 1:10 000-15 000 новорожденных. Избегание голодания и прием добавок карнитина могут спасти жизнь таким пациентам.
Нарушение метаболизма жирных кислот также лежит в основе гипогликемии натощак, связанной с ямайской рвотной болезнью, и отравления атрактилозидом и вальпроатом. При ямайской рвотной болезни незрелые плоды аки (Blighiasapida), содержащие водорастворимый токсин гипоглицин, вызывают рвоту, угнетение ЦНС и тяжелую гипогликемию. Гипогликемическая активность гипоглицина обусловлена его ингибированием глюконеогенеза, вторичным по отношению к его вмешательству в метаболизм ацил-КоА и карнитина, необходимых для окисления длинноцепочечных жирных кислот. Заболевание за редким исключением встречается только на Ямайке, где основным продуктом питания бедного населения являются фрукты. Зрелые плоды аки не содержат этого токсина.
Атрактилозид ингибирует окислительное фосфорилирование в митохондриях, препятствуя перемещению адениновых нуклеотидов, таких как АТФ, через митохондриальную мембрану. Атрактилозид является пергидрофенантреническим гликозидом, полученным из Atractylis gummifera. Данное растение встречается в бассейне Средиземного моря; употребление этого «чертополоха» связано с гипогликемией и синдромом, похожим на ямайскую тошноту. Подобное заболевание, отмеченное в Индии (острая токсическая энцефалопатия — гипогликемический синдром), м.б. вызвано употреблением личи. Личи содержит гипоглицин А и/или метиленциклопропилглицин, которые могут ингибировать окисление жирных кислот или глюконеогенез.
Противосудорожное ЛС вальпроат связано с побочными эффектами, преимущественно у младенцев, включающими синдром Рейе, низкий уровень карнитина в сыворотке крови и возможность развития гипогликемии при голодании.
Во всех этих условиях гипогликемия не связана с кетонемией и кетонурией.
- Острая алкогольная интоксикация. Печень метаболизирует алкоголь как предпочтительную энергию. Образование восстанавливающих эквивалентов во время окисления этанола изменяет НАДН: соотношение НАД, которое важно для определенных глюконеогенных стадий. В результате нарушается глюконеогенез и может возникнуть гипогликемия, если запасы гликогена истощаются из-за голодания или ранее существовавших аномалий метаболизма гликогена. У детей ясельного возраста, которые некоторое время голодали, даже небольшое количество алкоголя может ускорить эти явления. Гипогликемия быстро устраняется в/в введением глюкозы, что всегда следует учитывать при взятии образца крови для определения концентрации глюкозы ребенка, у которого изначально была кома или судороги. Возможность алкогольного отравления ребенка особенно велика, если накануне у родителей была вечеринка. Тщательный сбор анамнеза позволяет установить диагноз и избежать ненужной и дорогостоящей госпитализации и обследования.
- Отравление салицилатом. См. отдельную статью на сайте - просим Вас пользоваться формой поиска по сайту выше.
Как гипергликемия, так и гипогликемия встречаются у детей с интоксикацией салицилатами. Ускоренное использование глюкозы в результате увеличения секреции инсулина салицилатами и возможное вмешательство в глюконеогенез может способствовать развитию гипогликемии. Младенцы более восприимчивы, чем дети старшего возраста. Мониторинг уровня глюкозы в крови с соответствующей инфузией глюкозы в случае гипогликемии должен быть частью терапевтического подхода к салицилатной интоксикации в детстве. Иногда возникает кетоз.
- Недостаточность фосфоенолпируваткарбоксикиназы. Дефицит ограничивающего скорость глюконеогенного фермента фосфоенолпируваткарбоксикиназы связан с тяжелой гипогликемией при голодании и вариабельным началом после рождения. Гипогликемия может возникнуть в течение 24 ч после рождения, а нарушение глюконеогенеза аланина м.б. документально подтверждено in vivo. Печень, почки и миокард демонстрируют жировую инфильтрацию, и может возникнуть атрофия зрительного нерва и зрительной зоны коры. Гипогликемия м.б. глубокой. Уровни лактата и пирувата в плазме в норме, но может присутствовать умеренный метаболический ацидоз. Жировая инфильтрация разл. органов вызвана повышенным образованием ацетил-КоА, который становится доступным для синтеза жирных кислот. Диагноз этой редкой нозологической формы возможен лишь при определении активности фермента в биоптатах печени или при молекулярной диагностике.
Целесообразно исключить периоды голодания за счет частого приема пищи и рациона, богатого углеводами.
- Недостаточность пируваткарбоксилазы. См. отдельную статью на сайте - просим Вас пользоваться формой поиска по сайту выше.
8. Другие нарушения функции ферментов:
- Галактоземия (дефицит галактозо-1-фосфат-уридилтрансферазы). См. отдельную статью на сайте - просим Вас пользоваться формой поиска по сайту выше.
- Непереносимость фруктозы (дефицит фруктозо-1 -фосфатальдолазы). См. отдельную статью на сайте - просим Вас пользоваться формой поиска по сайту выше. Острая гипогликемия вызывается дефицитом фруктозо-1-фосфатальдолазы, которая подавляет гликогенолиз через систему фосфорилазы и глюконеогенез на уровне фруктозо-1,6-дифосфатальдолазы. Больные обычно учатся исключать фруктозу из своего рациона самостоятельно.
9. Дефекты транспортеров глюкозы:
- Дефицит транспортера глюкозы-1 в эритроцитах. В редких случаях обнаруживается, что у младенцев с судорожным расстройством низкие концентрации глюкозы в СМЖ на фоне нормального уровня ее в плазме. Концентрации лактата в СМЖ низкие, что свидетельствует о снижении гликолиза, а не о бактериальной инфекции, которая вызывает низкий уровень глюкозы в СМЖ с высоким содержанием лактата. Транспортер глюкозы в эритроцитах (GLUT-1) дефективный, что предполагает аналогичное нарушение и в ГМ, ответственном за клинические проявления. Кетогенная диета ослабляет судороги, предоставляя альтернативный источник энергии для ГМ, позволяющий избежать нарушения транспорта глюкозы.
- Дефицит транспортера глюкозы-2 в эритроцитах. У детей с гепатомегалией, непереносимостью галактозы и дисфункцией почечных канальцев (синдром Фанкони-Бикеля) наблюдается дефицит GLUT-2 плазматических мембран. Помимо печеночных и почечных канальцев, GLUT-2 также экспрессируется в [3-клетках ПЖЖ. Т.о., клинические проявления отражают нарушение высвобождения глюкозы из печени и ее канальцевой реабсорбции в почках, а также фосфатурию и аминоацидурию.
10. Системные заболевания. Некоторые системные расстройства связаны с гипогликемией у младенцев и детей. Неонатальный сепсис часто ассоциируется с гипогликемией, вероятно, в результате снижения потребления калорий с нарушением глюконеогенеза. Подобные механизмы могут применяться к гипогликемии, обнаруживаемой у детей с тяжелым истощением или с тяжелой мальабсорбцией. Гипервязкость с центральным гематокритом >65% связана с гипогликемией по крайней мере у 10-15% пострадавших младенцев.
Тропическая малярия связана с гиперинсулинемией и гипогликемией. Механизмы развития гипогликемии при СН и почечной недостаточности остаются неясными.
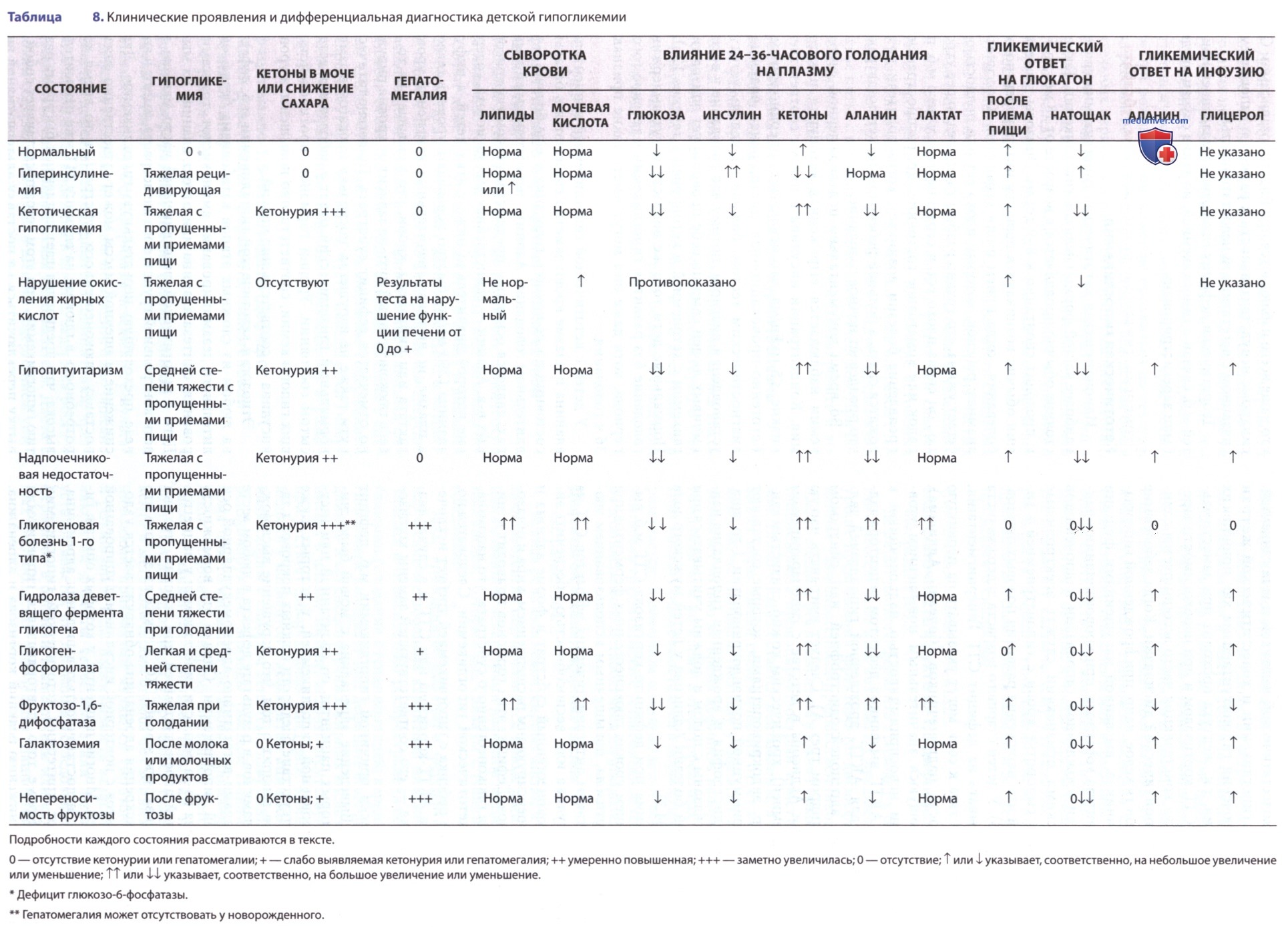
д) Диагностика и дифференциальная диагностика. В табл. 8 и на рис. 5 приведены соответствующие клинические и биохим. данные о распространенных детских расстройствах, связанных с гипогликемией. Тщательный и подробный анамнез важен для каждого подозреваемого или доказанного случая гипогликемии (см. табл. 7). Особые моменты, на которые следует обратить внимание, включают возраст, при котором возникли первые симптомы заболевания, связь с приемом пищи или недостаточной калорийностью рациона, а также семейный анамнез детей, перенесших гипогликемию, или необъяснимые смерти младенцев.
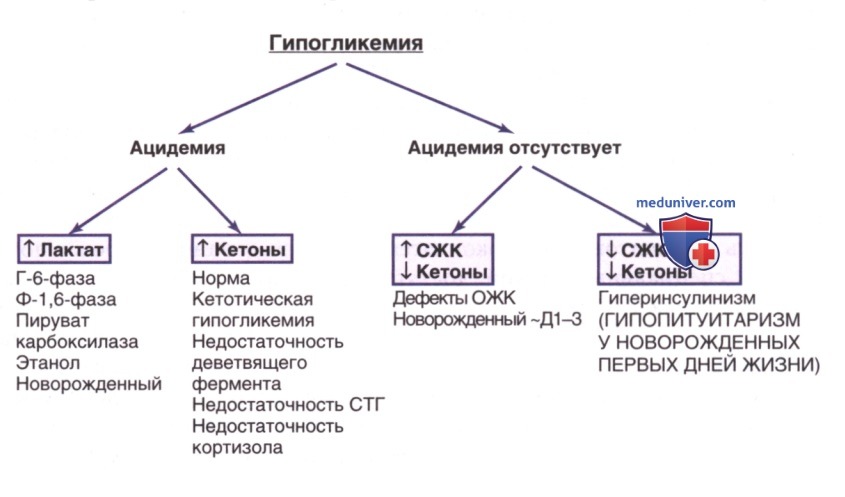
Рисунок 5. Алгоритм диагностики гипогликемии на основе энергетических реакций натощак. СЖК — свободные жирные кислоты; Ф-1,6-фаза — фруктозо-1,6-дифосфатаза; Г-6-фаза — глюкозо-6-фосфатаза; дефекты ОЖК — дефекты окисления жирных кислот; Д1-3 — 1-3-й дни жизни
На первой неделе жизни у большинства младенцев наблюдается преходящая форма неонатальной гипогликемии либо в результате недоношенности/ограничения в/утроб-ного развития, либо в результате рождения от матерей с СД. При отсутствии в анамнезе у матери СД, но наличии макросомии и признаков полнокровия, характерных для детей матери с СД, следует предположить возможность гиперинсулинемической гипогликемии младенчества, вероятно, в результате дефекта канала КАТФ (АуР), который является семейным или спорадическим. Снижение уровня β-гидроксибутирата, низкие СЖК и концентрации инсулина в плазме >5 мкЕд/мл или С-пептида >0,5 нг/мл при наличии доказанной гипогликемии подтверждают данный диагноз.
Наличие гепатомегалии должно вызывать подозрение на дефицит фермента, такого как глюкозо-6-фосфатаза, при болезни накопления гликогена (БНГ) I или др. БНГ; если в моче присутствует сахар, не снижающий глюкозу (напр., положительный результат по Clinitest, но отрицательный по Clinistix), наиболее вероятна галактоземия.
У мальчиков наличие микрофаллоса предполагает возможность гипопитуитаризма, который у детей обоего пола м.б. связан с холестатической желтухой. Наличие лицевого дефекта по срединной линии, такого как волчья пасть, также предполагает возможный гипопитуитаризм как причину гипогликемии из-за дефицита СТГ и/или кортизола. Высокий уровень настороженности и осведомленности о гипогликемии как о причине необычного поведения любого больного новорожденного требует определения уровня глюкозы у постели больного. Однако поскольку глюкометры имеют точность только ± 20%, любое значение глюкозы в крови <60 мг/дл должно быть подтверждено формальным лабораторным измерением, которое выполняется на образце крови, хранящемся в пробирке, которая предотвращает гликолиз, что может вызывают ложные низкие значения.
После периода новорожденности ключи к разгадке причины стойкой или рецидивирующей гипогликемии м.б. получены путем тщательного сбора анамнеза, физикального обследования и первоначальных лабораторных данных. Временная связь гипогликемии с приемом пищи может указывать на то, что нарушение связано с глюконеогенезом, если симптомы проявляются через 6 ч или после еды. Если гипогликемия возникает вскоре после еды, следует заподозрить и подтвердить или исключить гиперинсулинизм путем измерения β-гидроксибутирата, инсулина, С-пептида и СЖК в образце, в котором уровень глюкозы в крови <55 мг/дл. Необходимо учитывать АуД-формы гиперинсулинемической гипогликемии с измерением глюкозы, инсулина и аммиака и тщательным сбором семейного анамнеза.
Целесообразно измерение IGFBP-1; низкий при состояниях гиперинсулинизма и высокий при др. формах гипогликемии. Наличие гепатомегалии предполагает один из дефицитов ферментов в расщеплении гликогена или в глюконеогенезе, как показано в табл. 8. Отсутствие кетонемии или кетонурии на начальном этапе служит доказательством развития гиперинсулинизма или нарушения окисления жирных кислот. При большинстве др. причин гипогликемии, за исключением галактоземии и непереносимости фруктозы, кетонемия и кетонурия присутствуют во время гипогликемии при голодании. Во время гипогликемии необходимо получить сыворотку для определения субстратов, особенно глюкозы, β-гидроксибутирата, лактата и свободных жирных кислот, а также гормонов, особенно инсулина, С-пептида, кортизола, АКТГ и СТГ, с последующим повторным измерением глюкозы после в/м или в/в инъекции глюкагона, как указано в табл. 7. Табл. 8 суммирует интерпретацию результатов.
Гипогликемия с кетонурией у детей в возрасте от 18 мес до 5 лет, скорее всего, будет кетотической, особенно если отсутствует гепатомегалия. Прием токсина, в т.ч. алкоголя или салицилата, обычно выясняется из анамнеза. Также следует учитывать случайное или преднамеренное использование ЛС и ошибки в дозе ЛС. Если родители или др. лица, осуществляющие уход, имеют доступ к инсулину или стимуляторам секреции инсулина, следует учитывать мнимое расстройство (синдром Мюнхгаузена по доверенности) — высокие концентрации инсулина в образце с низкими концентрациями С-пептида подтверждают введение экзогенного инсулина. Преднамеренный или случайный прием ЛС, стимулирующих секрецию эндогенного инсулина, приведет к повышению концентрации как инсулина, так и С-пептида, и может потребоваться использование специальных лабораторных методов, позволяющих идентифицировать в-во, вызывающее нарушение.
Если анамнез позволяет предположить, но острых симптомов нет, 24-ч голодание под наблюдением обычно может спровоцировать гипогликемию и решить вопрос о наличии гиперинсулинизма или др. состояний (см. табл. 8). Такое голодание редко нужно продлевать до 36 ч, и только у детей старшего возраста. Оно противопоказано при подозрении на нарушение окисления жирных кислот. Следует рассмотреть др. подходы, такие как масс-тандемная спектрометрия или молекулярная диагностика, или и то и другое. Поскольку надпочечниковая недостаточность может имитировать кетотическую гипогликемию, уровни кортизола и АКТГ в плазме следует определять во время доказанной гипогликемии; усиление пигментации щек или кожи может указывать на первичную надпочечниковую недостаточность с повышенной активностью АКТГ (меланоцит-стимулирующего гормона). Низкий рост или снижение скорости роста могут дать ключ к разгадке гипофизарной недостаточности с участием СТГ, а также АКТГ.
Могут потребоваться окончательные тесты функции гипофиза и надпочечников, такие как тест стимуляции аргинином-инсулином на СТГ, IGF-1, IGFBP-1 и выброс кортизола.
При наличии гепатомегалии и гипогликемии предполагаемый диагноз ферментного нарушения м.б. поставлен на основании клинических проявлений, наличия гиперлипидемии, ацидоза, гиперурикемии, реакции на глюкагон при приеме пищи и натощак, а также реакции на инфузию разл. подходящих предшественников (см. табл. 7). В табл. 8 обобщены эти клинические данные и исследовательские подходы. Для окончательного диагноза гликогеноза может потребоваться молекулярная диагностика. У отдельных пациентов со всеми проявлениями гликогеноза обнаруживается нормальная активность ферментов. Такие окончательные исследования требуют специальных знаний, доступных только в определенных учреждениях.
е) Лечение. Предотвращение гипогликемии и ее последствий для развития ЦНС у новорожденных приобретает большое значение. Для новорожденных с гиперинсулинизмом, не связанным с СД у матери, может потребоваться субтотальная или фокальная панкреатэктомия, если только гипогликемию нельзя легко контролировать с помощью диазоксида в течение длительного времени, аналогов соматостатина (напр., октреотида) или сиролимуса. Изучаются др. новые подходы к лечению гиперинсулинемической гипогликемии.
Лечение острой симптоматической гипогликемии новорожденных или младенцев включает в/в введение 2 мл/кг 10% р-ра декстрозы («Глюкозы») в воде (D10W) с последующим непрерывным вливанием глюкозы в дозе 6-8 мг/кг в минуту с корректировкой скорости для поддержания уровня глюкозы в крови в пределах нормы. При наличии гипогликемических приступов некоторые рекомендуют болюсное введение 4 мл/кг D10W.
Лечение бессимптомной гипогликемии у младенцев из группы риска обычно включает энтеральное питание, а не парентеральное введение глюкозы. Если симптомы развиваются или гипогликемия сохраняется, несмотря на энтеральное питание, показано в/в введение глюкозы. Гель декстрозы («Глюкозы») (40%, 400 мг/кг), вводимый внутрь, м.б. альтернативой энтеральному кормлению, если грудное молоко или смесь недоступны.
Если необходимо, лечение стойкой неонатальной или младенческой гипогликемии включает увеличение скорости в/в инфузии глюкозы до 10-15 мг/кг в минуту или более. Для введения гипертонического 15-25% р-ра декстрозы («Глюкозы») могут потребоваться центральный или пупочный венозные катетеры. Если присутствует гиперинсулинизм, его следует лечить сначала диазоксидом, а затем аналогами соматостатина. Если гипогликемия не реагирует на в/в введение глюкозы с диазоксидом (максСД <15 мг/кг в сутки) и аналогов соматостатина, следует рассмотреть возможность частичной или почти полной панкреатэктомии. Такая операция должна проводиться в центрах с необходимым оборудованием и обученным персоналом, имеющим опыт проведения операций. Если возможно, хирургическому вмешательству должно предшествовать сканирование 18F-L-ДОФА для локализации поражения, которое затем, перед проведением операции, может дать хирургу рекомендации по клинически радикальной резекции.
Пероральный диазоксид 5-15 мг/кг в сутки в дробных дозах 2 р/сут может предотвратить развитие гиперинсулинемической гипогликемии, но может также вызвать гирсутизм, отек, тошноту, гиперурикемию, электролитные нарушения, увеличение костного возраста, дефицит IgG и, в редких случаях, гипертонию с длительным течением. Аналог соматостатина, октреотид, длительного действия, эффективен для контроля гиперинсулинизма, вызывающего гипогликемию у пациентов с нарушениями островковых клеток, включая генетические мутации в канале КАТФ и аденому островковых клеток. У новорожденных и младенцев младшего возраста глюкагон, вводимый путем непрерывной в/в инфузии 5 мкг/кг в час вместе с октреотидом, 20-50 мкг/кг в сутки п/к Q6-12H, могут поддерживать уровень глюкозы в крови, но они обычно используются в качестве временной меры перед частичной или более полной панкреатэктомией.
Возможные осложнения от приема октреотида включают замедленный рост из-за нарушения высвобождения СТГ, боль в месте инъекции, рвоту, диарею, нарушение функций печени (гепатит, холелитиаз) и некротический энтероколит; тахифилаксия к действию ЛП встречается чаще. Октреотид м.б. эффективен для лечения рефрактерной гипогликемии, несмотря на субтотальную панкреатэктомию.
Тотальная панкреатэктомия не является оптимальным методом лечения из-за рисков хирургического вмешательства, постоянного СД и экзокринной недостаточности ПЖЖ. Продолжение длительной медикаментозной терапии без резекции ПЖЖ при поддающейся контролю гипогликемии имеет смысл, поскольку со временем у некоторых детей происходит спонтанное разрешение гипогликемии, вызванной гиперинсулиниз-мом. Это должно быть сбалансировано с учетом риска повреждения ЦНС, вызванного гипогликемией, и токсичности ЛП.
ж) Прогноз. Прогноз благоприятный для новорожденных без симптомов с непродолжительной гипогликемией. После адекватного лечения гипогликемия рецидивирует у 10-15% младенцев. Рецидив более распространен.