Указанные формы протопорфирии генетически различны, но, по существу, имеют один и тот же фенотип. При ЭПП (АуР-заболевание) мутации FECH приводят к значительному дефициту FECH (последнего фермента в пути биосинтеза гема), в результате чего происходит накопление протопорфирина. ЭПП иногда называют эритропеченочной протопорфирией, хотя печень не вносит существенного вклада в продуцирование избыточного протопорфирина в неосложненных случаях. Порфирия Х-ПП описана совсем недавно.
При ХПС мутации ALAS2 приводят к усилению функции и избыточному продуцированию АЛК в костном мозге, где она метаболизируется с образованием избыточного количества протопорфирина.
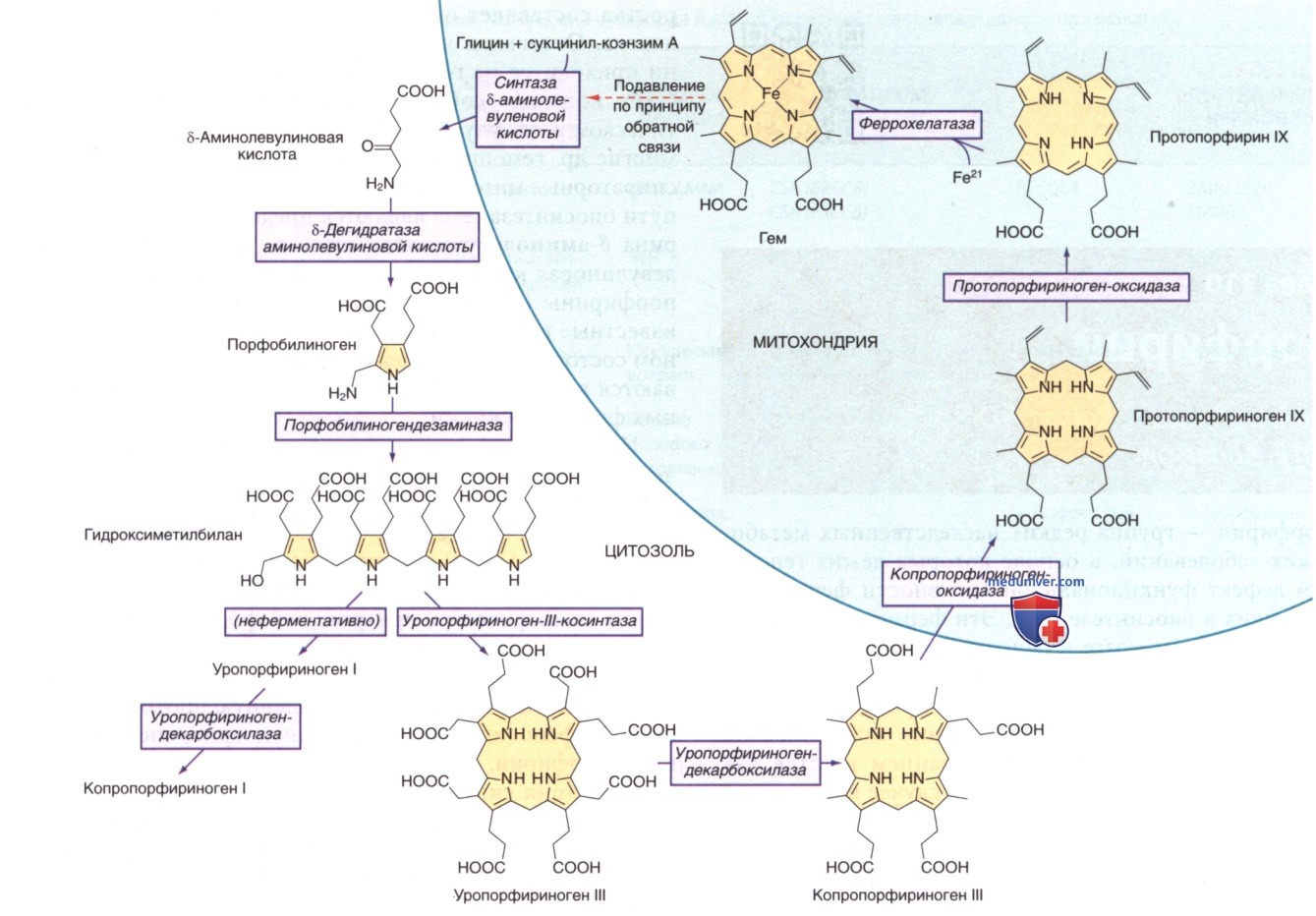
а) Этиология. При ЭПП дефицитным оказывается фермент феррохелатаза, который катализирует заключительный этап синтеза гема, а именно внедрение двухвалентного железа (Fe2+) в протопорфирин IX (см. рис. ниже). Фермент также называют гем-синтетазой или протогем-ферролиазой. Человеческий фермент является димером, и каждый гомодимер содержит кластер [2Fe-2S], который, вероятно, участвует в образовании мостиковых связей между гомодимерами. FECH находится во внутренней мембране митохондрий, и ее активный центр обращен к митохондриальному матриксу.
Ферменты и промежуточные продукты пути биосинтеза гема. Этот путь регулируется в печени конечным продуктом, гемом, в основном за счет подавления обратной связи (пунктирная стрелка)
Она м.б. связана с комплексом I митохондриальной цепи переноса электронов, а при окислении никотинамидадениндинуклеотида может происходить образование субстрата двухвалентного железа. FECH специфична для восстановленной формы железа, но может использовать др. металлы, такие как цинк и кобальт, а также др. дикарбоксилпорфирины. При ЭПП происходит накопление свободного протопорфирина вместо цинк-протопорфирина. Это означает, что образование последнего зависит от активности FECH in vivo.
Человеческий ген FECH расположен в хромосоме 18q21.3, имеет одну промоторную последовательность и содержит 11 экзонов. Были описаны две мРНК размером 1,6 и 2,5 кб, что можно объяснить использованием двух альтернативных сигналов полиаденилирования. Транскрипт большего размера чаще встречается в эритроидных клетках мышей, что предполагает эритроид-специфическую регуляцию FECH. При ЭПП зарегистрировано множество мутаций гена FECH, в т.ч. миссенс-мутации, нонсенс-мутации и сплайсинговые мутации, малые и большие делеции, а также инсерции.
Для того чтобы заболевание проявилось, необходимо унаследовать два аллеля, связанных со снижением активности FECH. Вышеописанное соответствует тому факту, что у пациентов с ЭПП показатели активности FECH составляют 15-20% от нормы. У большинства пациентов патогенетическая мутация 1-го аллеля FECH сочетается с распространенным вариантом мутации, поражающим др. аллель. Этот распространенный вариант аллеля FECH (IVS3-48T>C) сопровождается синтезом фермента в количествах ниже нормы, поскольку при этом происходит аберрантный сплайсинг мРНК, и такая мРНК разрушается под воздействием механизма нонсенс-опосредованного распада РНК.
Вариант FECH IVS3-48T>C сам по себе не вызывает развития заболевания, даже у гомозигот. В нескольких семьях были обнаружены две тяжелые мутации FECH без аллеля IVS3-48T>C.
В естественных условиях ЭПП с АуР-типом наследования наблюдают на кошачьих и мышиных моделях.
Х-ПП развивается на фоне делеций с усилением функции в последнем экзоне ALAS2. В результате этой мутации удаляются последние 10-20 аминокислот полипептида ALAS2, после чего фермент, по-видимому, становится более стабильным. В таких случаях в эритроцитах преобладает безметалловый протопорфирин, однако, поскольку активность FECH не отклоняется от нормы, доля цинкпротопорфирина больше, чем при классической ЭПП. На Х-ПП приходится ~2% случаев с фенотипом ЭПП в Европе и 10% случаев в Северной Америке.
В некоторых случаях ЭПП сопровождается миелодиспластическими синдромами и экспансией клона гематопоэтических клеток с делецией 1-го аллеля FECH или др. мутациями FECH. Для таких пациентов характерно позднее начало заболевания.
б) Эпидемиология. ЭПП чаще вызывает симптомы у детей, чем все остальные порфирии, но диагноз нередко устанавливают только во взрослом возрасте. Данное заболевание занимает третье место по распространенности среди всех порфирий, но точные показатели распространенности неизвестны (см. табл. 2). ЭПП описана в основном у представителей европеоидной расы, но встречается и среди представителей др. рас. Сплайс-вариант IVS3-48T>C распространен среди представителей европеоидной расы и японцев, но редко встречается у африканцев. Это объясняет низкие показатели распространенности заболевания среди выходцев из Африки.
в) Патология и патогенез. При ЭПП дефицит FECH наблюдается во всех тканях, но считается, что основным источником избыточного синтеза протопорфирина являются ретикулоциты костного мозга. Из них некоторое количество протопорфиринов выходит в плазму крови и переносится в кожу. Циркулирующие эритроциты больше не синтезируют гем и Hb, но содержат избыточное количество свободного протопорфирина, что также вносит вклад в развитие болезни. При Х-ПП, развивающейся в результате терминальных делеций в экзоне 11 ALAS2, все промежуточные продукты пути синтеза гема образуются в избыточных количествах и в конечном счете накапливаются в эритробластах костного мозга в виде протопорфирина.
Дефицита FECH при Х-ПП не наблюдается, так что этот фермент катализирует связывание некоторых количеств избыточного протопорфирина в хелатные комплексы с цинком. Кроме того, при Х-ПП описана транскрипция митоферрина, образовавшегося в результате аберрантного сплайсинга. Эта транскрипция ограничивает перенос железа в митохондрии. Печень функционирует скорее как экскреторный орган, чем как значимый источник избыточных количеств протопорфирина. Дефицит FECH в коже и печени может играть большую роль, поскольку данные исследований трансплантации тканей у мышей свидетельствуют о том, что фоточувствительность кожи и повреждение печени возникают только на фоне дефицита FECH в этих тканях.
У пациентов с ЭПП и Х-ПП максимальная чувствительность к свету определяется при длине волны 400 нм, что соответствует так называемой полосе Soret — узкой, наиболее интенсивной полосе максимального поглощения, характерной для протопорфирина и др. порфиринов. Поглощая свет, порфирины переходят в возбужденное состояние и высвобождают энергию в виде флуоресценции, синглетного кислорода и др.активных форм кислорода. Развивающееся в результате повреждение тканей сопровождается перекисным окислением липидов, окислением аминокислот, образованием поперечных сшивок в белках клеточных мембран, а также повреждением эндотелиоцитов капилляров. Такое повреждение может опосредоваться фотоактивацией системы комплемента и высвобождением гистамина, кининов и факторов хемотаксиса.
Повторные острые повреждения приводят к утолщению стенок сосудов и появлению периваскулярных отложений, состоящих из компонентов сыворотки. В верхних слоях дермы вокруг кровеносных сосудов наблюдают отложения аморфного материала, содержащего Ig, компоненты системы комплемента, гликопротеины, кислые гликазаминогликаны и липиды.
Существует мало свидетельств нарушения эритропоэза или гемолиза при ЭПП. Однако нередко встречается гипохромная микроцитарная анемия легкой степени и ретикулоцитоз. У некоторых пациентов отмечается накопление железа в эритробластах и кольцевидных сидеробластах костного мозга. Пониженные показатели насыщения трансферрина железом и показатели ферритина сыворотки ниже нормы или на нижней границе нормы указывают на дефицит железа. У пациентов с ЭПП следует тщательно оценивать состояние запасов железа в организме. При этом следует иметь в виду, что дефицит железа может привести к дальнейшему повышению уровней протопорфирина и ↑ риска холестаза.
У пациентов с ЭПП описан слабый ответ на применение ЛП железа внутрь. Объяснений данному факту не найдено. Некоторые пациенты отмечают ↑ фоточувствительности на фоне применения ЛП железа, но неизвестно, связано ли это с преходящим повышением уровней порфиринов на фоне коррекции дефицита железа и повышения активности эритропоэза. Данные сообщений указывают на то, что на фоне применения ЛП железа ↓ уровень протопорфирина и ↓ выраженность анемии, особенно у пациентов с Х-ПП.
У небольшой доли пациентов с ЭПП и Х-ПП развивается повреждение печени. Предполагают, что это связано с избыточным количеством протопорфирина, который не растворяется в воде и выделяется только через печень, а билиарная экскреция снижена в связи с холестазом. Некоторое количество протопорфирина реабсорбируется в кишечнике и подвергается энтерогепатической рециркуляции. На фоне холестаза избыточный протопорфирин, который накапливается в печени, может формировать в гепатоцитах кристаллические структуры и нарушать функцию митохондрий.
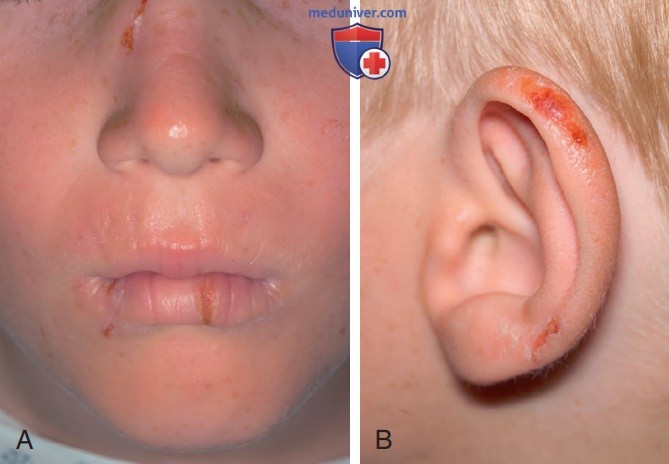
г) Клинические проявления. Симптомы кожной фоточувствительности манифестируют в детском возрасте в виде острой боли и зуда. Они нередко возникают после нескольких минут пребывания на солнце. После этого, если воздействие солнечных лучей продолжается, возникают покраснение и отек (рис. ниже). М.б. петехии и пурпура, но пузыри образуются в редких случаях. Отек может напоминать ангионевротический и солнечную крапивницу. Весной и летом симптомы обычно усугубляются. К возможным хроническим изменениям относятся лихенификация, кожные псевдовезикулы, губная борозда и изменения ногтей, но изменения пигментации и выраженные рубцы встречаются редко.
Эритропоэтическая протопорфирия (ЭПП): А — линейные эрозии латеральных поверхностей переносицы и нижней губы у пациента с ЭПП; В — эрозии, покрытые корочками в области завитка левой ушной раковины у пациента с ЭПП
Хотя физические признаки при ЭПП и Х-ПП м.б. незначительными, симптомы ухудшают качество жизни в значительно большей степени, чем при ПКП или ВП. Взаимосвязь между ЭПП, вызванной мутациями обеих аллелей FECH, и сезонной кератодермией ладоней не объяснена. Невропатия развивается только у некоторых пациентов с тяжелой печеночной недостаточностью. Мужчины с Х-ПП имеют более тяжелый фенотип с более высоким содержанием протопорфирина, чем большинство пациентов с ЭПП. У женщин с Х-ПП клинические проявления различаются: у некоторых симптомы отсутствуют или наблюдаются в легкой степени, а у др. присутствуют тяжелые симптомы, похожие на симптомы мужчин с Х-ПП. Такая вариативность симптомов у женщин, вероятно, является результатом случайной инактивации Х-хромосомы.
Что удивительно, несмотря на развитие осложнений со стороны печени или др. органов, уровни протопорфирина и симптомы фоточувствительности у многих пациентов остаются стабильными в течение многих лет. Факторы, которые приводят к обострению печеночной порфирии, при ЭПП и Х-ПП не играют никакой роли или их роль незначительна. В период беременности уровни протопорфирина эритроцитов могут ↓, а переносимость солнечного света — улучшаться. Этому факту объяснений не найдено.
д) Лабораторные признаки. При ЭПП значительно ↑ уровень протопорфирина в циркулирующих эритроцитах, практически 100% которого приходится на свободный (безметалловый) протопорфирин. При Х-ПП ↑ уровни цинк-протопорфирина и свободного протопорфирина, хотя последний все равно преобладает. Кроме того, ↑ уровни протопорфирина в костном мозге, плазме, желчи и фекалиях. При неосложненном течении ЭПП и Х-ПП уровни др. порфиринов и предшественников порфирина не отклоняются от нормы.
е) Диагностика и дифференциальная диагностика. Диагноз ЭПП подтверждается биохимически при обнаружении значительно повышенных концентраций общего протопорфирина в эритроцитах, который состоит преимущественно (не менее чем на 85%) из безметаллового протопорфирина и не образует комплексных соединений с цинком. При Х-ПП повышены как уровни свободного протопорфирина, так и уровни цинк-протопорфирина. В среднем при Х-ПП уровни общего протопорфирина в эритроцитах выше, а при ЭПП отличаются большей вариабельностью между пациентами. Возможно, это связано с различиями в степени тяжести множества зарегистрированных мутаций FECH.
При гомозиготных формах порфирий (кроме ВЭП), дефиците железа, отравлении свинцом, анемии на фоне хронических заболеваний, при состояниях, сопровождающихся гемолизом, а также при многих др. эритроцитарных нарушениях концентрация цинк-протопорфирина в эритроцитах ↑, при этом уровни безметаллового протопорфирина ↑ незначительно. Для измерений активности FECH необходимы клетки, содержащие митохондрии. Такой анализ не является широкодоступным.
Общая концентрация порфирина в плазме при ЭПП обычно ниже, чем при др. кожных порфириях, и может даже соответствовать норме. Порфирины плазмы при ЭПП особенно подвержены фоторазложению, поэтому при отборе пробы следует с особой осторожностью избегать воздействия света. Содержание порфиринов и их предшественников не повышено.
Для подтверждения мутаций FECH или ALAS2 и генетического консультирования настоятельно рекомендуется ДНК-анализ.
Жизнеугрожающая протопорфирическая гепатопатия характеризуется более значительным повышением концентраций протопорфирина в эритроцитах и плазме, повышенной светочувствительностью и либо хроническими нарушениями функции печени, либо быстро прогрессирующей печеночной недостаточностью. Предполагают, что предвестником этого состояния является увеличение уровней порфирина в эритроцитах и плазме крови выше исходных для данного пациента, но этот факт не подтвержден документально, поскольку у большинства таких пациентов не определяли должным образом исходные показатели уровня порфирина. В такой ситуации повышение уровней порфиринов, особенно копропорфирина, в моче объясняется нарушением функции печени.
ж) Осложнения. Имеется повышенный риск образования желчных камней, которые содержат протопорфирин и в некоторых случаях вызывают симптомы, требующие холецистэктомии. Протопорфирическая гепатопатия возникает у <5% пациентов с протопорфирией, в т.ч. у детей. Она может протекать хронически или стремительно прогрессировать, завершаясь летальным исходом от печеночной недостаточности. В некоторых случаях основным проявлением ЭПП становится заболевание печени. При Х-ПП заболевание печени может возникать более часто.
В одном сообщении, содержащем сведения о восьми семьях, у 17% пациентов имелись явные нарушения функции печени. На фоне протопорфирической гепатопатии может возникать острая боль в животе, указывающая на обструкцию ЖВП, и ненужная лапаротомия, проводимая с целью исключения этого состояния, может приводить к пагубным последствиям. Вносить свой вклад могут сопутствующие заболевания, сопровождающиеся нарушением функции печени, напр. вирусный гепатит, заболевания печени, индуцированные алкоголем, ЛС или оральными контрацептивами. Неизвестно, имеет ли значение дефицит железа. При гист. исследовании печени отчетливо видны отложения протопорфирина в виде включений в гепатоцитах и желчных капиллярах.
У пациентов с протопорфирической печеночной недостаточностью чаще всего имеются «нулевые мутации» FECH-аллель гипоэкспрессии IVS3-48T>C, но у некоторых пациентов м.б. два аллеля с тяжелыми мутациями FECH или ХСП, вызванная делециями 11-го экзона ALAS2. Вероятно, основным источником протопорфирина, даже у пациентов с ЭПП, сопровождающейся печеночной недостаточностью, является костный мозг.
з) Лечение. Следует избегать воздействия солнечных лучей, для этого рекомендуется носить одежду из плотной ткани. Систематический обзор вариантов лечения, включая прием бетакаротена («Бета-каротина»), цистеина (внутрь) и витамина С, не показал доказанной эффективности этих методов лечения. В одном отчете предполагалось, что высокие дозы циметидина эффективно снизили симптоматику у трех детей с ЭПП, но объективных клинических доказательств эффективности представлено не было.
Также м.б. полезны меры по созданию загара кожи. Для этого можно использовать узкополосную фототерапию УФ типа В (средневолновую). Двойные слепые плацебо-контролируемые исследования в США и Европе Афамеланотида, синтетического аналога а-меланоцитстимулирующего гормона, показали увеличение безболезненного пребывания на солнце и улучшение качества жизни у пациентов с протопорфирией. Указанный ЛП одобрен для использования взрослыми в Европе и ожидает утверждения FDA. Также планируется проведение исследований на детях.
Следует избегать приема ЛП или гормональных ЛП, нарушающих выделительную функцию печени, особенно пациентам с нарушениями функции печени. Кроме того, следует скорректировать дефицит железа, если он присутствует, особенно при Х-ПП. Рекомендуются добавки витамина D и вакцинация против гепатита А и В.
Лечение протопорфирной гепатопатии должно быть индивидуальным. Результаты лечения являются непредсказуемыми. Урсодезоксихолевая кислота м.б. в некоторой степени полезной на ранних стадиях. Холестирамин или активированный уголь могут ↓ энтерогепатическую циркуляцию протопорфирина, способствовать его экскреции с калом и ↓ содержание протопорфирина в печени. Может произойти самопроизвольное выздоровление, особенно при наличии обратимых причин дисфункции печени, таких как вирусный гепатит или злоупотребление алкоголем.
Для пациентов с тяжелой печеночной недостаточностью м.б. полезными (в т.ч. при трансплантации печени) такие методы лечения, как комбинированная терапия с плазмаферезом, переливание крови для коррекции анемии и подавления эритропоэза, в/в введение гемина для подавления продуцирования протопорфирина в эритроцитах и, возможно, в печени, прием урсодезоксихолевой кислоты, витамина Е и холестирамина.
У пациентов с протопорфирией (с заболеванием печени до или после трансфузии или трансплантации печени) может развиваться двигательная невропатия, напоминающая таковую при острой порфирии, иногда являющаяся обратимой. Искусственное освещение, напр. освещение операционной во время трансплантации печени или др. операций, может спровоцировать фотосенсибилизацию тяжелой степени с обширными ожогами кожи и брюшины, а также с повреждением циркулирующих эритроцитов.
Поскольку костный мозг продолжает продуцировать избыточное количество протопорфирина, даже после трансплантации может произойти рецидив заболевания печени. Тем не менее результаты сравнимы с трансплантацией печени при др. типах заболеваний. После трансплантации печени, при наличии подходящего донора, также должна рассматриваться трансплантация костного мозга.
и) Прогноз. Типичные пациенты с ЭПП имеют повышенную светочувствительность в течение всей жизни, но в остальном могут рассчитывать на полноценную долгую жизнь. Протопорфирия печени часто является жизнеугрожающим заболеванием, но встречается редко.
к) Профилактика и генетическое консультирование. Для профилактики возникновения симптомов рекомендуется избегать воздействия солнечного света. Для профилактики осложнений со стороны печени следует избегать ЛП, способных вызвать повреждение печени. Мнения о необходимости терапии ЛП железа расходятся, и в настоящее время этот вопрос исследуется.
При генетическом консультировании большое значение имеют исследования ДНК, проводимые с целью выявления мутаций FECH, гипоэкспрессии распространенного аллеля FECH с мутацией IVS3-48T>C или делеций 11-го экзона ALAS2. В случаях, когда развитие ЭПП связано с тяжелой мутацией FECH и наличием распространенного аллеля FECH с мутацией IVS3-48T>C, анализ ДНК супруга/супруги с целью определения наличия или, что более вероятно, отсутствия аллеля с гипоэкспрессией позволяет прогнозировать риск развития ЭПП у будущих детей. Состояние ЭПП может улучшиться в период беременности.