а) Абсанс приступы. Типичные абсанс приступы обычно начинаются в 5-8 лет и часто из-за своей краткости игнорируются родителями в течение многих месяцев, хотя они могут возникать до сотен раз в сутки. В отличие от фокальных приступов с нарушением осознания они не имеют ауры, обычно длятся всего несколько секунд и сопровождаются трепетанием век или закатыванием глаз вверх, но обычно не сопровождаются более яркими автоматизмами, наблюдаемыми при фокальных приступах с нарушением осознания (абсансный приступ может иметь простые автоматизмы, такие как чмоканье губ или ковыряние в одежде, и голова может минимально наклоняться вперед).
Абсансы не имеют постиктального периода и характеризуются немедленным возобновлением того, что пациент делал до приступа. Гипервентиляция в течение 3-5 мин может вызвать приступ и сопровождающие их 3 Гц спайк-медленноволновые разряды. Наличие миоклонии век при закрывании глаз (синдром Дживонса) и периорбитальных, периоральных или конечностных миоклонических рывков (последние называются миоклоническими абсансами) с типичными абсансами обычно предсказывает трудности в подборе медикаментозного лечения.
Ранние абсанс приступы (<4 лет) или лекарственная резистентность должны инициировать оценку дефекта транспортера глюкозы, который часто ассоциируется с низким уровнем глюкозы в СМЖ и отклонением при определении секвенирования гена транспортера.
Атипичные абсанс приступы связаны с миоклоническими компонентами и изменениями тонуса головы (падение головы) и тела, а также обычно более трудны для лечения. Они провоцируются сонливостью и обычно сопровождаются спайк-медленноволновыми разрядами частотой 1-2 Гц.
Юношеские абсансные приступы похожи на типичные абсансы, но возникают в более позднем возрасте и сопровождаются 4-6 Гц спайк-медленноволновыми, полиспайк- и медленноволновыми разрядами. Они обычно связаны с ювенильной миоклонической эпилепсией (см. ниже раздел «Доброкачественные генерализованные эпилепсии»).
б) Генерализованные моторные приступы. Наиболее распространенными генерализованными моторными приступами являются генерализованные тонико-клонические, которые м.б. либо преимущественно генерализованными (двусторонними), либо фокальными, либо двусторонними тонико-клоническими из одностороннего фокуса. При отсутствии фокального компонента приступ обычно начинается с потери сознания, а иногда с внезапного крика, закатывания глаз вверх и общего тонического сокращения с падением, апноэ и цианозом. В некоторых случаях тоническому застыванию предшествует клонический или миоклонический компонент.
За тонической фазой следует клоническая фаза, которая по мере прогрессирования приступа показывает замедление ритмических сокращений до тех пор, пока приступ не прекратится, обычно через 1-2 мин. За этим часто следует недержание мочи и постиктальный период. Последние обычно длятся от нескольких минут до нескольких часов с оглушением и постиктальной сонливостью, слабостью, атаксией, гипер- или гипорефлексией и головными болями. Существует риск аспирации и травмы.
Первая помощь включает укладку пациента на бок, очищение полости рта, если он открыт, ослабление тесной одежды или снятие украшений, а также мягкое вытягивание головы и по возможности освобождение ДП квалифицированным специалистом. Рот не должен быть насильно открыт посторонним предметом (это может привести к смещению зубов, вызывая аспирацию) или пальцем во рту (это может привести к серьезной травме пальца). Многие пациенты имеют единичные генетические генерализованные тонико-клонические приступы, которые м.б. связаны с интеркуррентным заболеванием или с причиной, которая не установлена. Генерализованные тонические, атонические и астатические приступы часто возникают при тяжелых генерализованных детских эпилепсиях.
Генерализованные миоклонические приступы могут возникать как при доброкачественных, так и при трудно поддающихся лечению генерализованных эпилепсиях (см. ниже разделы «Доброкачественные генерализованные эпилепсии» и «Тяжелые генерализованные эпилепсии»).
в) Доброкачественные генерализованные эпилепсии. Абсанс эпилепсия детства обычно начинается в среднем детском возрасте и у большинства пациентов проходит до наступления зрелого возраста. У ~25% пациентов также развиваются генерализованные тонико-клонические судороги, у половины до и у половины после начала абсансов. Доброкачественная миоклоническая эпилепсия младенческого возраста характеризуется возникновением миоклонических и др. приступов в течение первого года жизни с генерализованными 3 Гц спайк-медленноволновыми разрядами. Часто поначалу бывает трудно отличить этот тип от более тяжелых синдромов, но последующее наблюдение позволяет уточнить диагноз.
Фебрильный синдром плюс проявляется фебрильными приступами и множественными типами генерализованных приступов у нескольких членов семьи, и иногда разные люди в пределах одной семьи проявляют разл. генерализованные и фебрильные типы приступов.
Ювенильная миоклоническая эпилепсия (синдром Янца) является наиболее распространенной генерализованной эпилепсией у молодых людей, на долю которой приходится 5% всех эпилепсий. Она была связана с мутациями во многих генах, включая CACNB4; CLNC2; EJM2, 3, 4, 5, 6, 7, 9; GABRA1; GABRD; и Миоклонин1/EFHC1 (см. табл. 3). Как правило, этот синдром начинается в раннем подростковом возрасте с одного или нескольких из следующих проявлений: миоклонические судороги по утрам, часто заставляющие пациента ронять вещи, генерализованные тонико-клонические или клоно-тонико-клонические приступы при пробуждении и юношеские абсансы. Депривация сна, алкоголь (у пожилых пациентов) и фотостимуляция или реже некоторые когнитивные действия могут выступать в качестве преципитаторов.
На ЭЭГ обычно обобщенные от 4 до 5 Гц полиспайк-медленноволновые разряды. Существуют и др. формы генерализованной эпилепсии, такие как фотопароксизмальная эпилепсия, при которой генерализованные тонико-клонические, абсансные или миоклонические генерализованные приступы вызываются фото стимулами, такими как стробоскопические огни, просмотр телевизионных каналов и видеоигр. Могут возникать и др. формы рефлекторной (т.е. вызванной раздражителями) эпилепсии; связанные с ней приступы обычно носят генерализованный характер, хотя некоторые из них м.б. фокальными.
г) Тяжелые генерализованные эпилепсии. Тяжелые генерализованные эпилепсии связаны с трудноизлечимыми приступами и задержкой развития. Ранняя миоклоническая инфантильная энцефалопатия начинается в течение первых 2 мес жизни с тяжелыми миоклоническими приступами и подавлением вспышек на ЭЭГ. Это обычно вызвано врожденными нарушениями метаболизма, такими как некетотическая гипергликемия. Ранняя инфантильная эпилептическая энцефалопатия (синдром Отахара) имеет сходный возраст начала и изменения ЭЭГ, но проявляется тоническими приступами и обычно вызывается пороками развития ГМ или мутациями синтаксин-связывающего белка 1.
Термин «ранняя инфантильная эпилептическая энцефалопатия» (EIEE; англ. Early Infantile Epileptic Encephalopathy) также применяется, главным образом генетиками, к растущему числу др. генетических эпилептических энцефалопатий и эпилептических энцефалопатий развития, которые связаны с увеличением числа специфических генов с патогенными мутациями (см. табл. 3). Они могут проявляться или не проявляться как синдром Отахары, но все они имеют общую характеристику ранней эпилептической энцефалопатии. Напр., EIEE тип 4 синдром Отахара, вызванный мутациями синтаксин-связывающего белка 1. Тяжелая миоклоническая эпилепсия младенческого возраста (синдром Драве) начинается как фокальный фебрильный эпилептический статус или фокальные фебрильные приступы и позже проявляется как миоклонические и др. типы приступов.
Синдром Веста дебютирует в возрасте 2-12 мес и состоит из триады: инфантильных эпилептических спазмов, которые обычно возникают в кластерах (особенно при сонливости или при возбуждении), регрессии развития и типичной картины ЭЭГ, называемой гипсаритмией (см. рис. ниже). Гипсаритмия — высоковольтный, медленный, хаотический фон ЭЭГ с мультифокальными всплесками. Пациенты с криптогенным (иногда называемым идиопатическим, теперь называемым неизвестной этиологии) синдромом Веста имеют нормальное развитие до начала заболевания, тогда как пациенты с симптоматическим синдромом Веста имеют предшествующую задержку развития вследствие перинатальных энцефалопатий, ВПР, метаболических нарушений или др. этиологии.
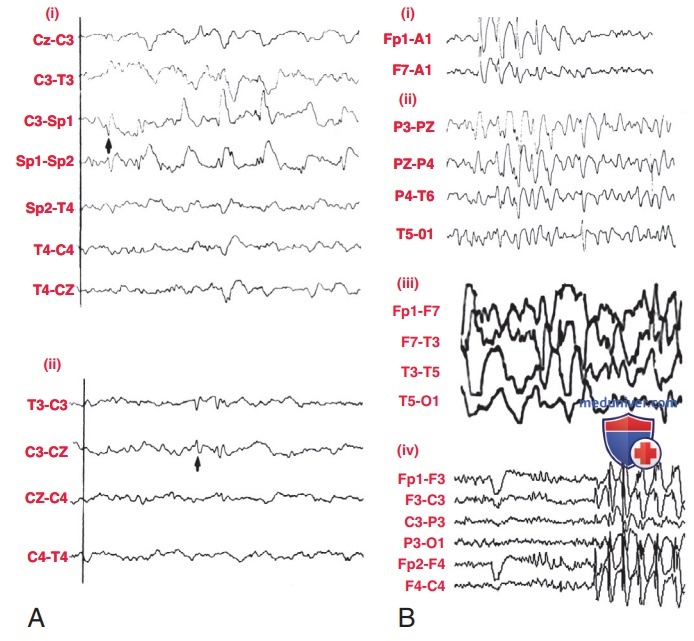
А — репрезентативная электроэнцефалография пациента с фокальными приступами: (i) спайковые разряды из левой височной доли (стрелка) у пациента со сложными фокальными приступами, вызванными мезиальным височным склерозом; (ii) левые Центрально-теменные спайки (стрелка), характерные для доброкачественной парциальной эпилепсии с центротемпоральными спайками; В — репрезентативные электроэнцефалограммы, ассоциированные с генерализованными приступами: (i) 3/с спайк-волновой разряд отсутствующих приступов с нормальной фоновой активностью; (ii) 1-2/с интериктальные медленные спайк-волны у пациента с синдромом Леннокса-Гасто; (iii) гипсаритмия с нерегулярной мультифокальной высоковольтной спайк-волновой активностью с хаотическим высоковольтным медленным фоном; (iv) ювенильная миоклоническая эпилепсия электроэнцефалография, показывающая спайк и волны 4-6/с, усиленные фотической стимуляцией
У мужчин синдром Веста также м.б. вызван мутациями гена ARX (часто связанными с неправильно развитыми гениталиями и аномалиями миграции коры ГМ). Синдром Веста, особенно в случаях неизвестной этиологии (криптогенные случаи, т.е. случаи, которые не являются симптомами метаболического или структурного нарушения ГМ), является показанием к НМП, поскольку задержка в диагностике на 3 нед или более может повлиять на долгосрочный прогноз. Спазмы часто игнорируются родителями и врачами, ошибочно принимаются за испуг, вызванный коликами или др. доброкачественными пароксизмальными синдромами.
Синдром Леннокса-Гасто обычно начинается в возрасте от 2 до 10 лет и состоит из триады: задержки развития, нескольких типов приступов, которые, как правило, включают атипичные абсансы, а также миоклонические, астатические и тонические приступы, а также специфические аномалии ЭЭГ. Тонические приступы возникают либо в бодрствовании (вызывая падения и травмы), либо во сне. Третий компонент — результаты ЭЭГ (см. рис. выше): спайк 1-2 Гц и медленные волны, всплески полиспайков во сне и медленный фон в бодрствовании. Пациенты обычно имеют тонические, миоклонические, атонические и др. типы приступов, вызывающих падения, и их трудно контролировать. У большинства формируются долгосрочные когнитивные нарушения и трудноизлечимые приступы, несмотря на многочисленные методы лечения.
У некоторых, но не у всех пациентов дебют идет с синдрома Отахары, далее развивается синдром Веста, а затем следует прогрессирование до синдрома Леннокса-Гасто. Миоклоническая астатическая эпилепсия (синдром Дузе) — сходный синдром, но более мягкий, чем синдром Леннокса-Гасто, который обычно не имеет тонических приступов или полиспайковых вспышек во сне. Прогноз более благоприятный, чем при синдроме Леннокса-Гасто. Др. синдромом, характеризующимся атоническими приступами в виде кивания головой, а также тоническими, клоническими и чувствительными к стимулам приступами, является синдром кивания, который наблюдается в некоторых африканских странах и часто ассоциируется с энцефалопатией, задержкой роста и разл. степенью когнитивного дефицита. Основной этиологией является вероятная аутоиммунная реакция на паразитического червя Onchocercavolvulus.
Прогрессирующие миоклонические эпилепсии — группа эпилепсий, характеризующихся прогрессирующим слабоумием и усилением миоклонических и др. приступов. Тип I, или болезнь Унферрихта-Лундборга, вызывается мутациями в гене цистатина В (CSTB), прогрессирует медленнее, чем др. типы, и обычно начинается в подростковом возрасте. Тип II, или болезнь Лафоры, может иметь начало в раннем детстве, но обычно начинается в подростковом возрасте, более быстро прогрессирует и обычно приводит к летальному исходу в течение 2-го или 3-го десятилетия.
Он м.б. связан с фоточувствительностью, проявляться периодическими кислотно-Шифф-«+»-включениями (тельца Лафоры) при биопсии мышц или кожи (в кл. эккринной потовой железы) и, как было показано, вызывается мутациями гена лафорина (ЕРМ2А) или малина (NHLRC1/ ЕРМ2В). Др. причины прогрессирующей миоклонической эпилепсии включают миоклоническую эпилепсию с «рваными красными волокнами» (MERRF, вызванные разл. мутациями в митохондриальной ДНК), сиалидоз I типа (вызванные мутациями в гене NEU1), нейрональный цероидный липофусциноз (лизосомные расстройства, вызванные мутациями в CLN1-CLN14 генов), тип 3 неврологической болезни Гоше (результат недостаточности глюкоцереброзидазы), дентаторубропаллидолюисовая атрофия (вызывается экспансией тринуклеотидных повторов в гене ATN1), миоклонус-синдром при почечной недостаточности [он же epilepsy, progressive myoclonic 4 (ЕРМ4), вызванный мутациями в гене SCARB2], прогрессирующая миоклонусная эпилепсия-атаксии (он же ЕРМ5, вызванная мутациями в гене PRICKLE1) и прогрессирующая миоклоническая эпилепсия Северного моря (он же ЕРМ6, вызванная мутациями в гене GOSR2).
Миоклоническая энцефалопатия при непрогрессирующих заболеваниях возникает при врожденных нарушениях ГМ, таких как синдром Ангельмана, и состоит из почти непрерывных и трудноизлечимых миоклонических и иногда др. приступов.
Синдром Ландау-Клефнера — редкое состояние предполагаемой аутоиммунной, но иногда и генетической (мутации GRIN2A) этиологии. Он характеризуется потерей языковых навыков и вербальной слуховой агнозией у ранее нормального ребенка. По крайней мере у 70% из них имеются сопутствующие клинические приступы, но у некоторых их нет. Приступы, когда они возникают, бывают нескольких типов, включая фокальные с сохраненным сознанием, очаговые с двусторонним тонико-клоническим компонентом, атипичные абсансы, очаговые с нарушенным осознанием и иногда миоклонические приступы.
Преобладают высокоамплитудные импульсно-волновые разряды, которые имеют тенденцию быть битемпоральными. На более поздних эволюционных стадиях этого состояния результаты ЭЭГ м.б. нормальными. Спайковые разряды всегда более заметны во время сна без быстрого движения глаз; т.о., ребенку, у которого подозревается синдром Ландау-Клефнера, необходимо провести ЭЭГ во время сна. Данные КТ и МРТ обычно нормальные. При родственном, но клинически отчетливом синдроме эпилепсии с непрерывными спайковыми волнами в медленноволновом сне (CSWS; англ. Continuous Spike Waves in Slow-Wave Sleep) разряды происходят в >85% медленноволнового сна, что называется электрическим статусом во сне (ESES; англ. Electrical Status Epilepticus in Sleep).
ESES может также возникать при синдроме Ландау-Клефнера, но при CSWS разряды обычно бывают фронтальными или генерализованными, а задержки обычно глобальными. Подход и терапия для этих двух синдромов схожи. Чаще всего из противоэпилептических ЛС применяется вальпроевая кислота, она используется в первую очередь для лечения клинических приступов и может помочь при афазии. Некоторые дети реагируют на клобазам, на комбинацию вальпроевой кислоты и клобазама или на леветирацетам. Для терапии афазии применяется диазепам на ночь (0,2-0,5 мг/кг внутрь перед сном в течение нескольких месяцев), часто используется в качестве терапии первой или второй линии, как и пероральные ГКС. Прием преднизолона внутрь начинают с 2 мг/кг в сутки в течение 1-2 мес, затем отлучают от груди в течение 1-3 мес.
Альтернативно ежемесячные в/в вливания метилпреднизолона в высокой дозе были использованы вместо пероральных ГКС. Длительная терапия часто необходима независимо от того, какой ЛП вызывает ответ пациента. Если судороги и афазия сохраняются после диазепама и стероидных проб, то следует рассмотреть курс в/в введения иммуноглобулинов, поскольку многие пациенты могут реагировать на это лечение. Необходимо начинать логопедическую терапию и поддерживать ее в течение нескольких лет, так как улучшение функции речи происходит в течение длительного периода.
Хорошо известны поддающиеся лечению метаболические эпилепсии. Пиридоксин-зависимая эпилепсия обычно представляет собой неонатальное или инфантильное (и редко детское) начало энцефалопатии с имеющимися данными об активности плода (приступов) в утробе матери. Существуют рецидивирующие фокальные моторные, генерализованные тонические приступы и миоклонус. Приступы прогрессируют до эпилептического статуса, если пиридоксин не назначен.
Диагноз подтверждается при наличии повышенного уровня α-аминоадипиновой полуальдегидной синтазы в плазме, моче, СМЖ и уровня пипеколиновой кислоты в плазме и СМЖ. Наличие либо гомозиготных, либо сложных гетерозиготных мутаций в аллелях ALDH7A1 (которые кодируют белок антиквитин) подтверждает диагноз. Применение пиридоксина внутрь по 100 мг/сут (применялись более высокие дозы, до 500-600 мг/сут) или в/в помогает купировать судороги.
Мутации гена PROSC также могут вызывать пиридоксин-зависимую эпилепсию. Пиридоксальфосфат-чувствительная неонатальная эпилептическая энцефалопатия [дефицит пиридокс(ам)ина 5’-фосфатоксидазы] [pyridox(am)ine 5’-phosphate oxidase (PNPO)] может проявляться аналогичным образом при отсутствии симптомов со стороны ЖКТ. При обследовании выявляется снижение уровня пиридоксальфосфата в ликворе при повышении уровня леводопы и 3-метокситирозина, а также снижение уровня гомованиловой кислоты и 5-гидроксииндолуксусной кислоты в ликворе. ЭЭГ может показывать паттерн подавления импульсов. Лечение заключается в энтеральном введении пиридоксальфосфата (до 50 мг/кг в сутки Q6H). Приступы, купирующиеся приемом фолиевой кислоты, могут также наблюдаться при неонатальной или инфантильной эпилептической энцефалопатии и трудноизлечимых припадках.
Некоторые из этих пациентов имеют диагностический профиль, аналогичный профилю пациентов с пиридоксин-зависимой эпилепсией, и их расстройство вызвано теми же мутациями гена, но отзывается на добавление фолиевой кислоты в дополнение к пиридоксину. Церебральный дефицит фолата, который также реагирует на высокие дозы фолиевой кислоты (1-3 мг/кг в сутки), может проявляться эпилепсией, умственной отсталостью, регрессией развития, дискинезиями и аутизмом. Уровень 5-метилтетрагидрофолата в СМЖ снижается при нормальном уровне фолиевой кислоты в плазме и эритроцитах крови.
Обычно имеются мутации в гене фолатного рецептора (FOLR1) или блокирующие аутоАТл к мембраносвязанным фолатным рецепторам сосудистого сплетения. Дефицит тетрагидробиоптерина с гиперфенилаланинемией или без нее может сопровождаться эпилепсией и симптомами, обусловленными дефицитом дофамина (паркинсонизм, дистония), норадреналина (аксиальная гипотония), серотонина (депрессия, бессонница, перепады ТТ) и фолиевой кислоты (образование миелина, кальцификация базальных ганглиев и судороги).
Лечение заключается в заместительной терапии тетрагидробиоптерином и предшественниками нейромедиаторов, начатой как можно раньше. Синдромы дефицита креатина обычно сопровождаются задержкой развития, судорогами, аутистическими особенностями и двигательными расстройствами и диагностируются с помощью секвенирования генов и аномальных уровней креатина в моче и гуанидиноуксусной кислоты и/или, особенно в случае дефицита транспортера креатина, отсутствием пика креатина на магнитно-резонансной спектроскопии ГМ. Полезно использовать моногидрат креатина и диетические ограничения.
Дефицит биотинидазы, проявляющийся задержкой развития, судорогами, атаксией, алопецией и кожной сыпью и часто связанный с прерывистым метаболическим ацидозом и органическим кислотным профилем молочной и пропионовой ацидемии, реагирует на применение биотина. Дефекты биосинтеза серина с его низким уровнем в плазме или аминокислотах СМЖ часто присутствуют у пациентов с врожденной микроцефалией, трудноизлечимыми приступами и психомоторной задержкой и реагируют на дополнительный серин и глицин.
Задержка развития, эпилепсия и неонатальный диабет вызваны активацией мутаций в чувствительных к АТФ калиевых каналах. ЛП сульфонилмочевины, блокирующие калиевый канал, лечат неонатальный диабет и, вероятно, также благоприятно влияют на симптомы ЦНС и купируют судороги. Гиперинсулинизм — синдром гипераммонемии, вызван активирующими мутациями глутаматдегидрогеназы, кодируемой геном GLUT1. У пациентов с гипогликемическими приступами после белковой еды наблюдается гипераммонемия (уровень аммиака 80-150 мкмоль/л). Они управляются с помощью комбинации ограничения белка, противоэпилептических ЛП и диазоксида (агониста калиевых каналов, который ингибирует высвобождение инсулина).
Синдром дефицита GLUT1 классически представляет собой инфантильную эпилепсию, задержку развития, приобретенную микроцефалию и сложные двигательные расстройства.
Он вызывает нарушение транспорта глюкозы в ГМ, которое обычно диагностируется с помощью генетического тестирования, или обнаружения низкого содержания лактата и глюкозы СМЖ, или низкого отношения глюкозы СМЖ к глюкозе сыворотки крови (<0,4). Симптомы болезни обычно реагируют на кетогенную диету.
Мутации переносчика тиамина при остром заболевании базальных ганглиев часто сопровождаются судорогами и чувствительны к добавкам биотина и тиамина. Дефицит транспортера рибофлавина также может проявляться в виде приступа в дополнение к обычным симптомам нервно-мышечной слабости (полиневропатии); его лечат добавлением высоких доз рибофлавина.
Видео первая и неотложная помощь при эпилепсии (судорогах, эпилептическом припадке)