Гемофилия А (дефицит фактора VIII) и гемофилия В (дефицит фактора IX) являются наиболее распространенными и серьезными видами наследственных дефицитов факторов свертывания крови. Клиническая картина при гемофилии А и гемофилии В практически идентична. Гемофилия С — это нарушение свертывания крови, связанное со снижением уровня фактора XI. Сниженные уровни контактных факторов (фактор XII, высокомолекулярный кининоген и прекал-ликреин) связаны со значительной пролонгацией АЧТВ; но не связаны с кровоизлияниями, как обсуждалось в отдельной статье на сайте - просим Вас пользоваться формой поиска по сайту выше.
Др., менее распространенные виды дефицитов факторов свертывания крови кратко обсуждаются ниже в последующих подразделах статьи.
а) Дефицит факторов VIII или IX (гемофилия А или В). Дефицит факторов VIII и IX являются наиболее распространенными формами тяжелого наследственного нарушения свертывания крови.
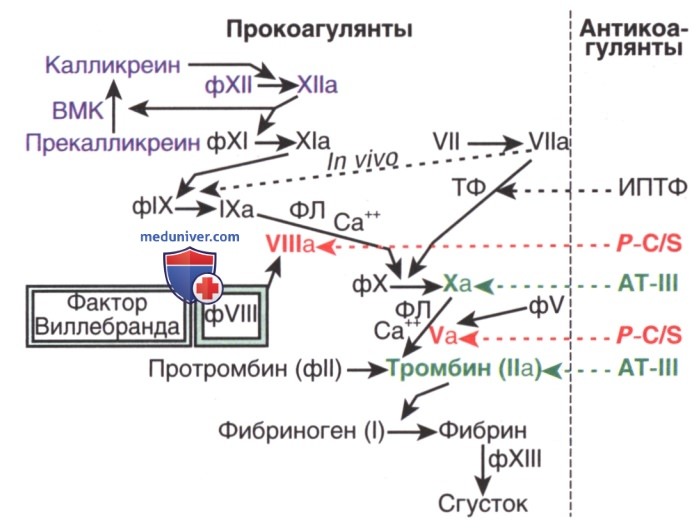
1. Патофизиология. Факторы VIII и IX участвуют в комплексе, необходимом для активации фактора X. Вместе с фосфолипидами и кальцием они образуют теназный комплекс, или комплекс, активирующий фактор X. На рис. ниже показан процесс свертывания крови, происходящий в пробирке, где фактор X активируется либо комплексом факторов VIII и IX, либо комплексом тканевого фактора и фактора VII. In vivo комплекс из фактора VIIa и тканевого фактора активирует фактор IX для инициации процесса свертывания крови. В лабораторных условиях ПТВ измеряет активацию фактора X фактором VII и поэтому находится в норме у пациентов с дефицитом фактора VIII или фактора IX.
Каскад свертывания крови с последовательной активацией и амплификацией (усилением) образования сгустка. Многие из факторов (ф) активируются факторами свертывания, расположенными в каскаде над ними. Активированные факторы обозначаются добавлением буквы а. Справа на рис. показаны основные антикоагулянты и участки, которые они регулируют: ингибитор пути тканевого фактора (ИПТФ) регулирует тканевой фактор (ТФ); фактор VIIa, протеин С и протеин S (P-C/S) регулируют факторы VIII и V; а антитромбин III (АТ-III) регулирует фактор Ха и тромбин (фактор IIа). Пунктирная линия показывает, что in vivo ТФ и фактор VIIa активируют оба фактора IX и X, но in vitro измеряется только активация фактора X. Неактивированный фактор VIII, будучи связанным со своим белком-носителем, фактором Виллебранда, защищен от инактивации протеином С. Когда тромбин, или фактор Ха, активируют фактор VIII, он становится несвязанным с фактором Виллебранда, в этот момент он может совместно с фактором IХа участвовать в активации фактора X в присутствии фосфолипидов (ФЛ) и кальция (теназный комплекс). Фактор ХIIIа перекрестно сшивает фибриновый сгусток и тем самым повышает его стабильность. Прекалликреин, высокомолекулярный кининоген (ВМК) и фактор XII изображены синим цветом, поскольку они не играют физиол. роли в коагуляции, хотя и оказывают влияние на показатели времени свертывания при определении активированного частичного тромбопластинового времени
После травмы на начальном этапе гемостатического каскада происходит образование тромбоцитарной пробки вместе с образованием фибринового сгустка, который предотвращает дальнейшее кровотечение. При гемофилии А и В образование фибринового сгустка замедляется и не является стабильным. Недостаточное образование тромбина приводит к невозможности образования фибринового сгустка с плотно сшитыми волокнами, поддерживающего тромбоцитарную пробку. У пациентов с гемофилией медленно образуется мягкий, рыхлый сгусток крови. Если кровотечение происходит в закрытом пространстве, напр. в суставе, остановить его можно с помощью тампонады. При открытых ранах, тампонада которых невозможна, обильное кровотечение может привести к значительной кровопотере.
Образовавшийся сгусток м.б. рыхлым, а во время физиол. лизиса сгустка или при минимальной новой травме может произойти повторное кровотечение.
2. Клинические проявления. Ни фактор VIII, ни фактор IX не проникают через плаценту; симптомы кровотечения могут наблюдаться с рождения или возникать у плода. Только 2% новорожденных с гемофилией страдают от в/черепных кровоизлияний, у 30% младенцев мужского пола с гемофилией наблюдается кровотечение при обрезании. Т.о., при отсутствии «+» семейного анамнеза (30% случаев гемофилии А возникает в результате спонтанной мутации) гемофилия у новорожденного может остаться недиагностированной. Очевидные симптомы, такие как склонность к образованию кровоподтеков, в/м гематомы и гемартрозы, обнаруживаются, когда ребенок начинает ходить.
Кровотечение из небольших травматических рваных ран полости рта (разрыв уздечки во рту) может сохраняться в течение нескольких часов или дней и может вынудить родителей обратиться за МП. Даже среди пациентов с тяжелой формой гемофилии только у 90% имеются признаки усиления кровотечения к 1-му году жизни. Хотя кровотечение может возникать в любой части тела, отличительной чертой кровотечения при гемофилии является гемартроз. Кровоизлияния в суставы м.б. вызвано легкой травмой; многие гемартрозы происходят спонтанно.
Самые ранние суставные кровоизлияния чаще всего появляются в голеностопе. У детей старшего возраста и подростков также часто встречаются гемартрозы коленных и локтевых суставов. В то время как ранние кровоизлияния в суставы у ребенка распознаются только после обширного отека и скопления жидкости в суставе, дети старшего возраста часто способны обнаружить у себя кровотечение раньше, чем врач. Дети жалуются на чувство тепла и покалывания в суставе, что является первым признаком раннего кровоизлияния в сустав. Повторные эпизоды кровотечения в один и тот же сустав у пациента с тяжелой гемофилией могут привести к появлению сустава-мишени. Из-за патологических изменений в суставе рецидивирующее кровотечение может стать самопроизвольным.
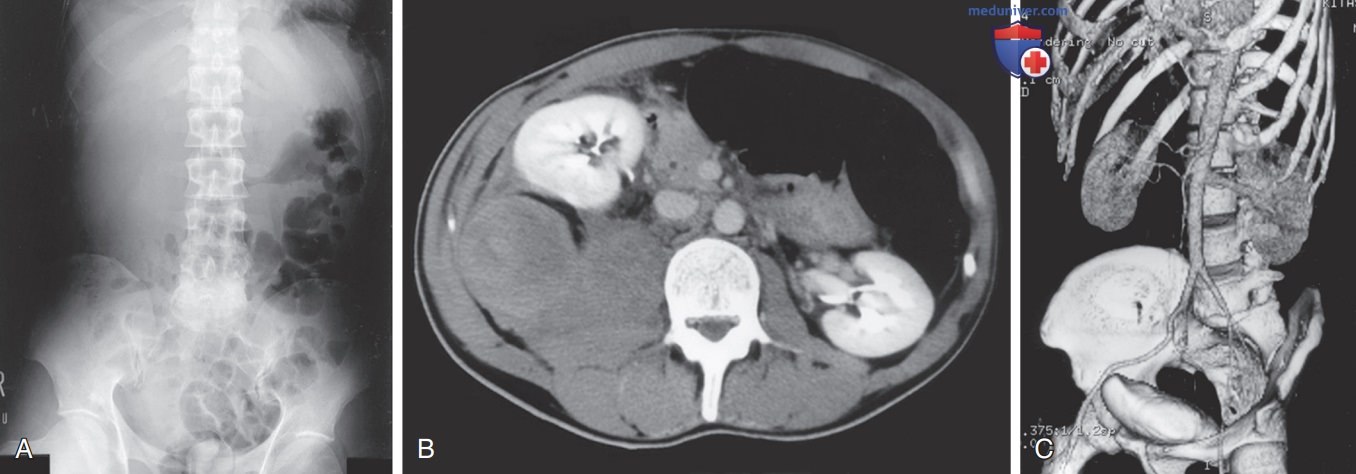
Хотя большинство мышечных кровоизлияний клинически очевидны из-за локализованных болей или отеков, кровоизлияние в подвздошно-поясничную мышцу требует отдельного упоминания. У пациента может произойти кровоизлияние больших объемов в подвздошно-поясничную мышцу на грани гиповолемического шока, при этом может ощущаться только реперкуссионная боль в неопределенной области паха. Бедро удерживается в согнутом положении и повернуто внутрь из-за воспаления подвздошно-поясничной мышцы. Диагноз ставится на основе клинического симптома невозможности разгибания бедра, но должен быть подтвержден УЗИ или КТ (рис. ниже).
Массивная гематома в области подвздошно-поясничной мышцы у пациента с гемофилией В. Мужчина 38 лет с выраженным дефицитом фактора IX (гемофилия В) поступил по поводу прогрессирующей боли и чувствительности в правом нижнем отделе брюшной полости. У него наблюдалось простудное заболевание с сильным кашлем и потерей аппетита на протяжении ~1 нед; А — рентгенограмма брюшной полости показывает псоас-симптом Образцова1 справа и смещение толстой кишки, содержащей газ, влево; В — компьютерная томография показывает массивную гематому в области правой подвздошно-поясничной мышцы, приводящую к передней транслокации правой почки; С — реконструированное трехмерное изображение более четко показывает транслокацию почки и расширенные, но неповрежденные крупные сосуды. Эти данные помогают при проведении диагностики, т.к. прогрессирующая боль в правом нижнем отделе брюшной полости может точно симулировать клинику острого аппендицита. Кровоизлияние было успешно устранено с помощью заместительной терапии фактором IX в течение 1 нед без каких-либо рецидивов. У пациента отсутствовали ингибиторы к фактору IX
Угрожающее жизни кровотечение у пациента с гемофилией вызвано кровоизлиянием в жизненно важные структуры (ЦИС, ВДП) или потерей крови (внешняя травма, ЖКК или кровоизлияние в подвздошно-поясничную мышцу). При угрожающих жизни кровоизлияниях необходимо оперативное лечение концентратом фактора свертывания крови. Если травма головы вызывает настолько серьезные опасения, что необходима рентгенологическая оценка, то перед рентгенологической оценкой следует провести заместительную терапию концентратом фактора свертывания.
Угрожающие жизни кровоизлияния требуют заместительной терапии с целью достижения уровня, соответствующего содержанию фактора в нормальной плазме крови (100 МЕ/дл, или 100%).
У пациентов с легкой формой гемофилии, имеющих уровень фактора VIII или фактора IX >5 МЕ/дл, обычно не наблюдаются самопроизвольные кровоизлияния. У таких пациентов может наблюдаться длительное кровотечение после стоматологических процедур, операций или травмы средней степени тяжести, но диагноз м.б. поставлен только, когда они достигнут более старшего возраста.
3. Лабораторные исследования и диагностика. При пониженном уровне фактора VIII или фактора IX результаты лабораторных исследований свидетельствуют о пролонгированном АЧТВ. При тяжелой форме гемофилии показатели АЧТВ обычно в 2-3 раза превышает верхнюю границу нормы. Результаты др. скрининговых тестов механизма гемостаза (количество тромбоцитов, время кровотечения, ПТВ, тромбиновое время) находятся в норме. Если у пациента отсутствует ингибитор фактора VIII или IX, смешивание нормальной плазмы с плазмой пациента приводит к коррекции значения АЧТВ. Специфический анализ на факторы VIII и IX подтвердит диагноз гемофилии. Если коррекция не происходит при смешивании нормальной плазмы с плазмой пациента, то это может указывать на присутствие ингибитора.
У 25-35% пациентов с гемофилией, получающих инфузии фактора VIII или фактора IX, может появиться специфическое АТл против соответствующего фактора. Эти АТл направлены против активного коагуляционного сайта и называются ингибиторами. У таких пациентов следует провести количественный анализ титра АТл в единицах Бетезда*.
P.S. * Одна единица Бетезда — БЕ/мл (Bethesda Unit, или BU/ml — в англоязычной транскрипции) определяет уровень ингибитора в плазме крови пациента, который приводит к 50% снижению активности фVIII (или фIХ) в смеси равных объемов плазмы донора и пациента после 2 ч инкубации при температуре 37 °C.
4. Дифференциальная диагностика. У младенцев первых месяцев жизни с тяжелыми симптомами кровотечения следует провести ДД тяжелой тромбоцитопении; тяжелых нарушений функции тромбоцитов, таких как синдром Бернара-Сулье (Bernard, Jean; Soulier, Jean Pierre) и тромбастения Гланцмана (Glanzmann, Eduard); болезни Виллебранда (Willebrand, Erik Adolf von) 3-го типа (тяжелой степени); а также дефицита витамина К.
5. Генетика и классификация. Гемофилия встречается ~у 1:5000 мужчин, причем у 85% наблюдается дефицит фактора VIII и у 10-15% — дефицит фактора IX. Гемофилия не имеет расовой предрасположенности и отмечается во всех этнических группах. Тяжесть гемофилии классифицируется на основании исходного уровня фактора VIII или фактора IX у пациента, поскольку уровни факторов обычно коррелируют с тяжестью симптомов кровотечения. По сути, 1 ME каждого фактора определяется как то количество фактора в 1 мл нормальной плазмы, которое соответствует стандарту, установленному ВОЗ; т.о., 100 мл нормальной плазмы содержат 100 МЕ/дл (100% активность) каждого фактора.
Для удобства обсуждения мы используем термин «% активности» для обозначения процентного содержания в нормальной плазме (100% активность). Концентраты факторов также соотносятся с международным стандартом ВОЗ, поэтому терапевтические дозы обычно указываются в ME. Тяжелая форма гемофилии характеризуется уровнем активности специфического фактора свертывания крови <1%, а кровотечение часто бывает самопроизвольным.
У пациентов с умеренной формой гемофилии уровни фактора составляют 1-5%, и обычно кровотечение возникает при легкой травме. У пациентов с легкой формой гемофилии уровень фактора составляет >5%, может пройти много лет, прежде чем это нарушение будет диагностировано, а кровотечение возникает обычно только при серьезной травме. Гемостатический уровень фактора VIII составляет >30-40%, а фактора IX >25-30%. Нижняя граница уровней факторов VIII и IX у ЗЛ составляет 50%.
Гены факторов VIII и IX расположены вблизи точки окончания длинного плеча Х-хромосомы и, следовательно, наследуются как Х-сцепленные признаки. У большинства пациентов с гемофилией отмечается пониженный уровень белка фактора свертывания крови; у 5-10% пациентов с гемофилией А и 40-50% пациентов с гемофилией В наблюдается дисфункциональный белок. У 45-50% пациентов с тяжелой формой гемофилии А встречается та же мутация, при которой происходит инверсия внутри гена фактора VIII, что приводит к полной остановке производства белка. Эта мутация м.б. обнаружена в крови пациентов или носителей, а также в амниотической жидкости с помощью молекулярных методов.
У афроамериканцев часто встречается др. гаплотип фактора VIII, т.о., повышенное образование ингибиторов у афроамериканцев м.б. обусловлено этим отличием. Т.к. в основе дефицитов фактора VIII или фактора IX лежат многие разные генетические причины, большинство случаев гемофилии классифицируются в зависимости от показателей активности фактора VIII или фактора IX. У новорожденных показатели фактора VIII м.б. неестественно повышены из-за ответа острой фазы, вызванного процессом рождения. В результате такого искусственного повышения у пациента с легкой формой гемофилии может наблюдаться нормальный или почти нормальный уровень фактора VIII. У пациентов с тяжелой формой гемофилии уровень фактора VIII не определяется. Вместе с тем уровень фактора IX у новорожденных физиол. низок.
Если в семье есть случаи тяжелой формы гемофилии, то неопределяемый уровень фактора IX является диагностическим признаком тяжелой гемофилии В. У некоторых пациентов с умеренным дефицитом фактора IX наличие гемофилии м.б. подтверждено только через несколько недель жизни.
Из-за инактивации Х-хромосомы у некоторых женщин-носителей гемофилии А или В наблюдается снижение фактора VIII или фактора IX до уровня, достаточного, чтобы вызвать легкие нарушения свертывания крови. Уровни этих факторов должны быть установлены у всех известных или потенциальных носителей для определения необходимости лечения в случае операции или клинического кровотечения.
Поскольку фактор VIII циркулирует в плазме крови совместно с фактором Виллебранда, соотношение фактора VIII и фактора Виллебранда иногда используется для диагностики носителей гемофилии, но результаты м.б. л/п или л/о. Специфические генетические мутации должны быть по возможности определены у пробанда, а результаты должны использоваться при тестировании др. членов семьи, которые подвергаются риску либо развить гемофилию, либо быть носителями.
6. Лечение. Правильный подход к лечению гемофилии заключается в ранней адекватной терапии. При легком или умеренном кровотечении показатели фактора VIII или фактора IX должны быть повышены до гемостатического уровня в диапазоне 35-50%. При угрожающих жизни или обширных кровотечениях целевая доза должна обеспечивать уровень 100% активности.
Расчет дозы рекомбинантного фактора VIII или рекомбинантного фактора IX производится следующим образом:
- Доза рекомбинантного фактора VIII (ME) = % желаемой активности (повышение уровня фактора VIII) х МТ (кг) х 0,5.
- Доза рекомбинантного фактора IX (ME) = % желаемой активности (повышение уровня фактора IX в плазме крови) х МТ (кг) х 1,3.
Коррекция для фактора VIII рассчитывается в зависимости от объема распределения фактора VIII. Коррекция для фактора IX основана на объеме распределения и наблюдаемом повышении уровня рекомбинантного фактора IX в плазме крови после инфузии.
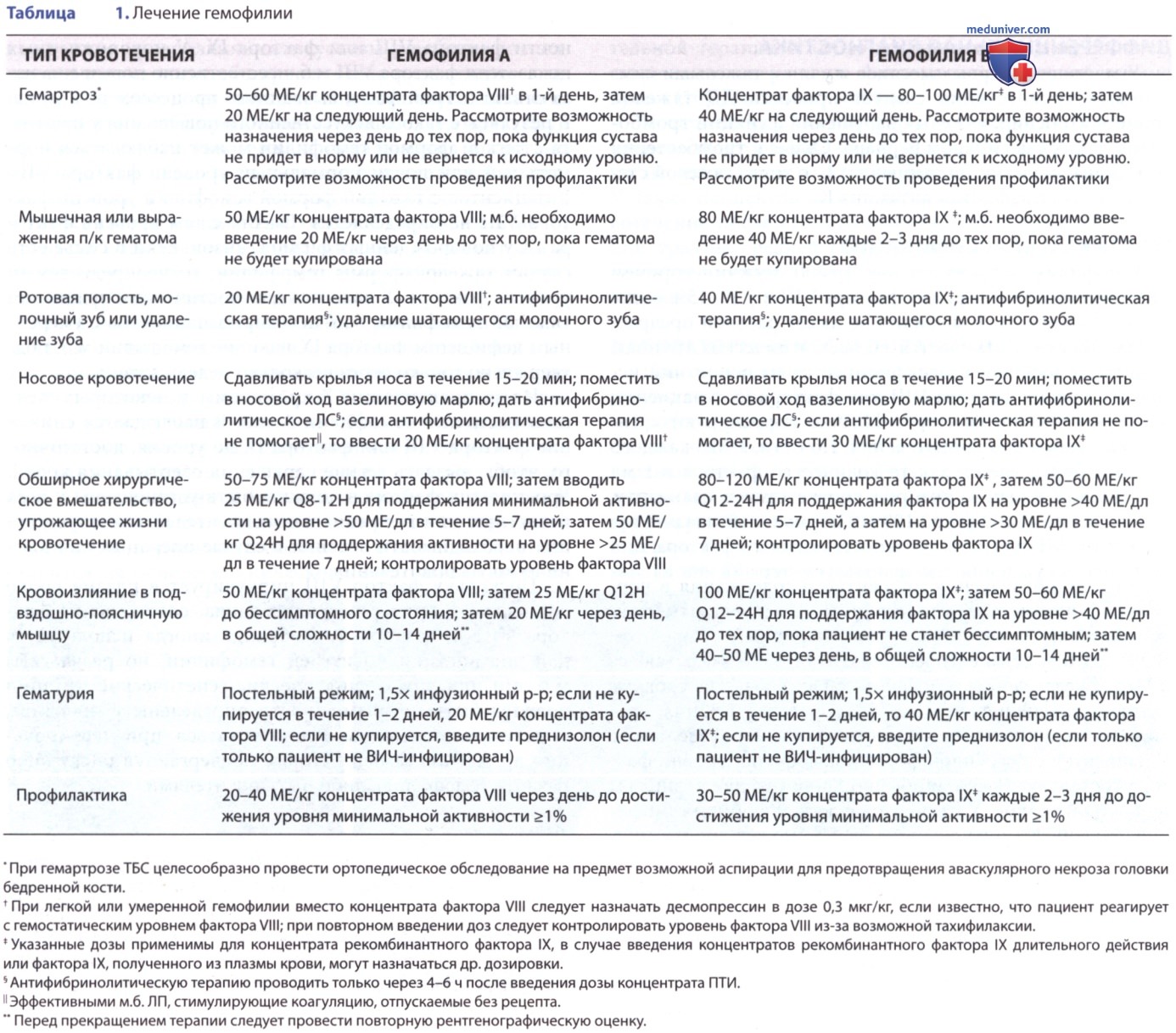
В табл. 1 представлены результаты лечения некоторых распространенных типов кровотечений у пациентов с гемофилией.
Благодаря наличию рекомбинантных ЛП заместительной терапии, стандартом оказания МП большинству детей с тяжелой формой гемофилии является профилактика, направленная на предотвращение самопроизвольных кровотечений и ранних деформаций суставов. В дополнение к имеющимся в настоящее время рекомбинантным факторам, разрабатываются ЛП, позволяющие увеличить период полувыведения из плазмы и снизить иммуногенность гемостатических факторов. Исследование, сравнивающее профилактику с агрессивными эпизодическими методами лечения, свидетельствует о преимуществе профилактики в предотвращении инвалидизирующих заболеваний суставов. При появлении суставов-мишеней часто проводится «вторичная» профилактика.
В случае легкой формы гемофилии А, фактор VIII, эндогенно продуцируемый пациентом, м.б. высвобожден при введении десмопрессина («Десмопрессина ацетата»). У пациентов с умеренным или тяжелым дефицитом фактора VIII накопленные уровни фактора VIII в организме недостаточны, и лечение десмопрессином неэффективно. Для лечения пациентов с легкой формой гемофилии А также можно использовать концентрированную интраназальную форму десмопрессина («Десмопрессина ацетата»), но не дозировку, предназначенную для лечения энуреза или для заместительной терапии после операции в области гипофиза. Доза составляет 150 мкг (1 впрыскивание) для детей МТ <50 кг и 300 мкг (2 впрыскивания) для детей и подростков МТ >50 кг.
В большинстве мед. центров проводятся исследования уровня фактора VIII, достигаемого после инфузионного введения десмопрессина. Десмопрессин не эффективен при лечении гемофилии с дефицитом фактора IX, но является эффективным и относительно менее дорогостоящим средством при лечении дефицита фактора VIII умеренной степени тяжести.
В настоящее время проводятся предварительные испытания генной терапии дефицита фактора IX с некоторыми обнадеживающими первичными результатами. Кровотечение из слизистой оболочки может потребовать дополнительного назначения антифибринолитика, такого как аминокапроновая кислота или транексамовая кислота.
Биспецифическое гуманизированное моноклональное АТл эмицизумаб может связывать активированный фактор IX и фактор X, восстанавливая т.о. функциональную активность активированного фактора VIII у пациентов с гемофилией (как у пациентов с наличием ингибиторов фактора VIII, так и у пациентов без них). Профилактические п/к инъекции эмицизумаба 1 р/нед могут снизить частоту кровотечений у пациентов с ингибиторами фактора VIII или без них.
7. Профилактика. В настоящее время многим пациентам на протяжении всей жизни проводится профилактика для того, чтобы предотвратить самопроизвольные кровоизлияния в суставы. Национальная гемофильная ассоциация рекомендует рассматривать профилактику как оптимальную терапию для детей с тяжелой формой гемофилии.
Обычно такие схемы лечения начинают проводить после 1-го или 2-го кровоизлияния в сустав. Маленьким детям часто требуется постановка центрального катетера для обеспечения венозного доступа. Такие схемы лечения являются дорогостоящими, но очень эффективными в предотвращении или значительном снижении степени тяжести заболевания при патологии суставов; осложнения включают в себя катетер-ассоциированную инфекцию и тромбоз. Лечение обычно проводится каждые 2-3 дня для поддержания измеримого уровня фактора свертывания крови в плазме крови (1-2%) при анализе непосредственно перед следующей инфузией (уровень минимальной остаточной активности).
Доступны более новые ЛП длительного действия, содержащие фактор IX, которые можно вводить реже, 1 р/нед или через неделю. Вопрос о том, следует ли продолжать профилактику в зрелом возрасте, еще недостаточно изучен.
При развитии умеренной артропатии для предотвращения будущих кровотечений потребуется более высокий уровень факторов свертывания крови в плазме крови. Детям более старшего возраста, которым не проводится первичная профилактика, часто назначается вторичная профилактика при появлении сустава-мишени.
8. Поддерживающая терапия. Самая простая рекомендация родителям состоит в том, что их ребенок должен избегать травм, но этот совет не подходит для активных детей и подростков. Малыши, которые уже начали ходить, активны, им все интересно, и они легко травмируются. К числу эффективных мер относятся упреждающий контроль, включая использование автомобильных сидений, ремней безопасности и велосипедных шлемов, а также предотвращение поведения с высокой степенью риска. Мальчикам более старшего возраста следует рекомендовать не заниматься агрессивными контактными видами спорта, но все не так просто. Мальчики с тяжелой формой гемофилии часто страдают от кровоизлияний и при отсутствии известных им травм.
Ранняя психосоциальная интервенция помогает семье найти баланс между чрезмерной защитой ребенка и вседозволенностью. Пациентам с гемофилией следует избегать приема ацетилсалициловой кислоты («Аспирина») и др. НПВП, влияющих на функцию тромбоцитов. Ребенок с нарушением свертывания крови должен получить соответствующие прививки против гепатита В, даже если рекомбинантные вакцины не заражены трансфузионно-ассоциированными заболеваниями. Пациенты, подвергающиеся воздействию ЛП, полученных из плазмы крови, должны периодически проходить обследование на гепатиты В и С, ВИЧ и нарушения функции печени.
9. Хронические осложнения. К числу долгосрочных осложнений гемофилии А и В относятся хроническая артропатия, появление ингибитора либо фактора VIII, либо фактора IX, а также риск инфекционных трансфузионно-ассоциированных заболеваний. Несмотря на то что инвазивный, или профилактический, подход к лечению снизил проблему хронической артропатии, эти проблемы не были устранены полностью.
В прошлом хроническая артропатия была основным видом длительной нетрудоспособности, ассоциированной с гемофилией. Естественная динамика гемофилии до лечения представляет собой повторяющиеся кровоизлияния в определенные суставы, в т.ч. кровоизлияния в один и тот же сустав (сустав-мишень). У маленьких детей сустав легко расширяется, и большой объем крови может заполнять сустав до тех пор, пока не будет применена тампонада или не начнется терапия. После кровоизлияния в сустав лейкоциты высвобождают в суставную полость протеолитические ферменты, а гемовое железо индуцирует пролиферацию макрофагов, что приводит к воспалению синовиальной оболочки.
Синовиальная оболочка уплотняется и образует в суставе выросты «на ножках», которые чувствительны к защемлению и могут привести к дальнейшим кровоизлияниям. Хрящевая поверхность эрозируется, и в конечном итоге может даже произойти обнажение кости, в результате чего возможно сращение суставов. У пациентов более старшего возраста с прогрессирующей артропатией, кровоизлияние в сустав-мишень с его утолщенной синовиальной оболочкой вызывает сильную боль, поскольку в суставе м.б. мало места для крови. Как только появляется сустав-мишень, пациенту обычно назначают краткосрочную или долговременную профилактику, чтобы предотвратить развитие артропатии и уменьшить воспаление.
- Образование ингибиторов. Инфузия ЛП, содержащего дефицитный фактор свертывания крови, может инициировать иммунный ответ у пациентов с дефицитом фактора VIII или фактора IX. Ингибиторы — это АТл, направленные против фактора VIII или фактора IX, которые блокируют свертывающую активность. Отсутствие коррекции кровотечения при соответствующей заместительной терапии, как правило, является первым признаком наличия ингибитора. Реже ингибиторы обнаруживаются во время планового контрольного скрининга на наличие ингибиторов. Ингибиторы выявляются у 25-35% пациентов с гемофилией А, но только у 2-3% пациентов с гемофилией В, у многих из которых вырабатывается неактивный дисфункциональный белок, что делает их менее восприимчивыми к иммунному ответу.
Введение высокоочищенного фактора IX или рекомбинантного фактора IX, по-видимому, увеличивает частоту появления ингибиторов, некоторые из них вызывают анафилаксию.
Многие пациенты, у которых есть ингибитор, теряют его при продолжении регулярных инфузий. У др. пациентов при последующих инфузиях наблюдается более высокий титр АТл и, возможно, таким пациентам потребуется пройти программу десенсибилизации (индукции иммунной толерантности), в ходе которой высокие дозы фактора VIII при гемофилии А или фактора IX при гемофилии В вводятся с целью насыщения АТл и создания условий для развития толерантности в организме. У некоторых пациентов программы иммунной толерантности к фактору IX способствовали появлению нефротического синдрома. Пациентам с высокими титрами ингибиторов, для которых программы иммунной толерантности оказались неэффективными, в качестве альтернативной терапии назначались ритуксимаб, ГКС и др. иммуносупрессивные средства.
В случае неудачной десенсибилизации эпизоды кровотечения лечатся либо с помощью рекомбинантного фактора VIIa, либо с помощью активированных концентратов протромбинового комплекса (с активностью, шунтирующей ингибиторы к фактору VIII). Эти ЛП во многих случаях обходят ингибитор, но могут увеличить риск тромбоза. Еще одним вариантом для пациентов с ингибиторами м.б. эмицизумаб. Некоторым пациентам с низкими титрами ингибитора могут назначаться высокие дозы фактора VIII во время эпизода кровотечения. Для лечения пациентов с ингибиторами необходимо направить их в мед. центр, который оказывает МП многим подобным пациентам и предлагает комплексную программу лечения гемофилии. Согласно некоторым данным, риск появления ингибиторов м.б. выше при использовании рекомбинантного фактора по сравнению с фактором, полученным из плазмы, однако необходимы дальнейшие исследования.
Раньше ЛП, полученные из плазмы крови, заражали большое количество пациентов с гемофилией гепатитами В и С, а также ВИЧ. В эпоху рекомбинантных продуктов риск заражения такими инфекциями должен быть минимальным, однако пациенты должны проходить соответствующую вакцинацию против гепатита В. Пациенты, подвергающиеся воздействию продуктов крови, должны находиться под наблюдением на предмет трасфузионноассоциированных инфекций. В отчетах также указывается на передачу варианта болезни Крейтцфельда-Якоба (Creutzfeldt, Hans Gerhard; Jakob, Alfons Maria) пациентам, получающим терапевтическую плазму, и может потребоваться исследование пациентов с гемофилией на предмет передачи прионов через концентраты фактора, полученного из плазмы.
10. Комплексный подход. МП пациентам с гемофилией лучше всего оказывается в центрах комплексного ухода за больными гемофилией. Такие центры занимаются обучением пациентов и членов их семей, а также профилактикой и лечением осложнений гемофилии, в т.ч. хронических заболеваний суставов, формирования ингибиторов, а также инфекций (напр., гепатита В или С и ВИЧ). К работе в таких центрах привлекается команда врачей, медсестер, ортопедов, физиотерапевтов и психосоциальных работников. Обучение по-прежнему имеет важнейшее значение при лечении гемофилии, поскольку пациенты, которым проводится профилактика, м.б. менее «опытными» в распознавании эпизодов кровотечения, чем дети предыдущих поколений.
б) Дефицит фактора XI (гемофилия С). Дефицит фактора XI — это аутосомный дефицит, ассоциированный с симптомами кровотечения легкой или умеренной степени тяжести. Он часто встречается у евреев-ашкенази, но может встречаться и во многих др. этнических группах. В Израиле 1-3:1000 являются гомозиготами по дефициту фактора XI.
Тенденция к кровотечениям не настолько выражена, как при дефиците фактора VIII или фактора IX. Кровотечение, связанное с дефицитом фактора XI, не коррелирует с количеством фактора XI. У некоторых пациентов с тяжелой формой дефицита могут наблюдаться незначительные симптомы во время обширной операции или же симптомы могут вообще отсутствовать. Поскольку фактор XI усиливает выработку тромбина и приводит к активации активируемого тромбином ингибитора фибринолиза, хирургическое кровотечение более выражено на участках с высокой фибринолитической активностью, таких как полость рта. За исключением случаев, когда пациент ранее перенес операцию без кровотечения, следует рассмотреть возможность проведения предоперационной заместительной терапии в зависимости от вида хирургического вмешательства.
В США нет одобренного к использованию концентрата фактора XI, поэтому врач должен использовать свежезамороженную плазму.
Кровотечение во время малого хирургического вмешательства можно контролировать с помощью локального давления. Пациенты при удалении зубов могут получать лечение ингибитором фибринолиза, таким как аминокапроновая кислота или транексамовая кислота, и такая терапия будет эффективна, при этом заместительная терапия плазмой будет применяться только в случае кровотечения. У пациента с гомозиготным дефицитом фактора XI АЧТВ часто бывает более пролонгированным, чем у пациентов с тяжелым дефицитом фактора VIII или фактора IX. Вызывает удивление парадоксальное сочетание меньшего количества клинических симптомов и более пролонгированного АЧТВ. Но это происходит потому, что фактор VIIa может активировать фактор IX in vivo. Дефицит фактора XI м.б. подтвержден специфическими тестами на определение активности фактор XI.
Плазменные инфузии в дозе 1 МЕ/кг обычно повышают активность фактора в плазме на 2%. Т.о., инфузия свежезамороженной плазмы 10-15 мл/кг приведет к повышению уровня фактора XI в плазме до 20-30%, что обычно достаточно для контроля умеренного кровотечения.
Для достижения более высоких уровней фактора XI необходимы частые инфузии плазмы. Поскольку период полувыведения фактора XI обычно составляет >48 ч, поддержание приемлемого уровня фактора XI как правило не является сложным.
При дефиците фактора XI редко развивается хроническое кровоизлияние в сустав, и для большинства пациентов этот дефицит представляет угрозу только во время обширных операций, кроме случаев, когда у пациента имеется второй дефект гемостаза (напр., болезнь Виллебранда).
в) Дефициты контактных факторов (нарушения, не связанные с кровотечением). Дефицит «контактных факторов» — фактора XII, прекалликреина и высокомолекулярного кининогена — приводит к пролонгированию АЧТВ, но не вызывает симптомов кровотечения. Поскольку эти контактные факторы функционируют на стадии инициации внутренней системы свертывания реагентом, используемым для определения АЧТВ, тест АЧТВ при их дефиците заметно пролонгирован. Т.о., возникает парадоксальная ситуация, при которой АЧТВ чрезвычайно пролонгировано без каких-либо клинических признаков кровотечения.
Очень важно, чтобы пациенты с такими данными были хорошо осведомлены о том, что означает дефицит фактора свертывания крови в их случае, т.к. они не нуждаются в лечении, даже при обширных операциях.
г) Дефицит фактора VII. Дефицит фактора VII — это редкое аутосомное нарушение свертывания крови, которое обычно выявляется только в гомозиготном состоянии. Степень тяжести кровотечения варьирует от легкой до тяжелой с гемартрозами, самопроизвольными в/черепными кровоизлияниями и кожно-слизистыми кровотечениями, особенно носовыми кровотечениями и меноррагией. Пациенты с этим дефицитом имеют значительно пролонгированное ПТВ, но нормальные показатели АЧТВ. Тесты на фактор VII показывают заметное снижение активности фактора VII. Поскольку период полувыведения фактора VII из плазмы составляет 2-4 ч, терапия свежезамороженной плазмой затруднена и часто осложняется перегрузкой жидкостью. Промышленно выпускаемый концентрат рекомбинантного фактора VIIa эффективен при лечении пациентов с дефицитом фактора VII.
д) Дефицит фактора X. Дефицит фактора X — это редкое (по оценкам, 1:1 млн) аутосомное заболевание различной степени тяжести. Дефицит легкой степени тяжести приводит к кожно-слизистым и посттравматическим кровотечениям, тогда как дефицит тяжелой степени приводит к самопроизвольным гемартрозам и в/черепным кровоизлияниям. Дефицит фактора X является результатом либо количественного дефекта, либо производства дисфункциональных молекул. Снижение уровня фактора X связано с пролонгацией как ПТВ, так и АЧТВ. У пациентов с наследственным дефицитом фактора X уровень фактора X м.б. повышен с помощью либо свежезамороженной плазмы, либо концентрата протромбинового комплекса. Период полувыведения фактора X составляет ~30 ч, а его объем распределения аналогичен объему распределения фактора IX. Т.о., 1 ед/кг увеличит плазменный уровень фактора X на 1%.
Хотя такая проблема редко встречается у педиатрических пациентов, системный амилоидоз м.б. связан с дефицитом фактора X, возникающим в результате адсорбции фактора X на амилоидном белке. В условиях амилоидоза трансфузионная терапия часто не является эффективной из-за быстрого клиренса фактора X.
е) Дефицит протромбина (фактора II). Дефицит протромбина обусловлен либо выраженным снижением уровня протромбина (гипопротромбинемия), либо функциональной недостаточностью протромбина (диспротромбинемия). Лабораторные исследования у гомозиготных пациентов показывают пролонгированные ПТВ и АЧТВ. Тесты на фактор II (протромбин) показывают значительное снижение уровня протромбина. В младенчестве часто наблюдаются кожно-слизистые кровотечения, а посттравматические кровотечения возникают в более позднем возрасте. Пациентам проводится терапия свежезамороженной плазмой, либо, реже, концентратами протромбинового комплекса. Свежезамороженная плазма эффективна при дефиците протромбина, потому что период полувыведения протромбина составляет 3,5 дня.
Введение 1 МЕ/кг свежезамороженной плазмы повышает активность протромбина в плазме крови на 1%. Приобретенный дефицит фактора II может наблюдаться при небольшом проценте волчаночных антикоагулянтов и обычно ассоциирован с выраженным кровотечением.
ж) Дефицит фактора V. Дефицит фактора V является АуР-нарушением свертывания крови с легкой или умеренной степенью тяжести геморрагических проявлений, которое также называют парагемофилией. Гемартрозы наблюдаются редко. Наиболее частыми симптомами являются кожно-слизистые кровотечения и гематомы. У женщин часто встречается меноррагия тяжелой степени. Лабораторные исследования показывают пролонгированные АЧТВ и ПТВ. Специфические тесты на фактор V показывают снижение уровня фактора V. Свежезамороженная плазма — это единственный доступный в настоящее время терапевтический продукт, содержащий фактор V. Фактор V быстро теряется во время хранения свежезамороженной плазмы. Пациентам с тяжелой формой дефицита фактора V назначаются инфузии свежезамороженной плазмы по 10 мл/кг Q12H.
Переливание тромбоцитов также является одним из вариантов лечения, поскольку тромбоциты содержат фактор V, что, возможно, объясняет отсутствие кровотечений, наблюдаемых у многих пациентов с дефицитом фактора V. В редких случаях у пациента с «-» семейным анамнезом кровотечения наблюдается приобретенное АТл к фактору V.
з) Комбинированный дефицит факторов V и VIII. Комбинированный дефицит факторов V и VIII возникает как вторичное нарушение из-за отсутствия в/клеточного транспортного пути, который отвечает за транспортировку факторов V и VIII из эндоплазматического ретикулума в аппарат Гольджи с участием LMAN1 и MCFD2. Это объясняет парадоксальный дефицит двух факторов, один из которых кодируется на хромосоме 1, а др.-на Х-хромосоме. Симптомы кровотечения часто более слабые, чем при гемофилии А, и их лечат с помощью свежезамороженной плазмы в качестве заместительной терапии как фактора V, так и фактора VIII.
и) Дефицит фибриногена (фактора I). Врожденная афибриногенемия — это редкое АуР заболевание, характеризующееся отсутствием фибриногена. У пациентов с афибриногенемией кровотечение наблюдается не так часто, как у пациентов с гемофилией, а также у них редко встречаются гемартрозы. У пациентов, страдающих этим заболеванием, в неонатальном периоде могут наблюдаться ЖКК или гематомы после естественных родов. В дополнение к выраженной пролонгации ПТВ и АЧТВ увеличивается и тромбиновое время. При отсутствии коагулопатии потребления не поддающийся измерению уровень фибриногена является диагностическим критерием. В дополнение к количественному дефициту фибриногена сообщалось о ряде дисфункциональных аномалий фибриногена (дисфибриногенемия). В редких случаях у пациентов с дисфибриногенемией наблюдается тромбоз.
Концентрат человеческого фибриногена является коммерчески доступным для терапии эпизодов кровотечений у больных афибриногенемией. Поскольку период полувыведения фибриногена из плазмы составляет 2-4 дня, терапия свежезамороженной плазмой или криопреципитатом тоже эффективна. Гемостатический уровень фибриногена составляет >60 мг/дл. Каждый пакет криопреципитата содержит 100-150 мг фибриногена. Некоторые клинические тесты на фибриноген ингибируются высокими дозами гепарина. Т.о., оценка значительно пролонгированного тромбинового времени, ассоциированного с низким уровнем фибриногена, должна проводиться вместе с определением рептилазного времени. Пролонгированное рептилазное время подтверждает, что функциональные уровни фибриногена низки, а гепарин отсутствует.
к) Дефицит фактора XIII (фибринстабилизирующего фактора, или дефицит трансглутаминазы). Поскольку фактор XIII отвечает за перекрестное сшивание фибрина для стабилизации фибринового сгустка, симптомы позднего кровотечения вторичны по отношению к нестабильности сгустка. Как правило, если у пациентов была травма в первый день, то кровоподтек или гематома появляются на следующий день. Клинические симптомы включают легкие кровоподтеки, позднее отделение пуповинного остатка через 4 нед у новорожденных, плохое заживление ран и повторные самопроизвольные аборты у женщин. Были описаны редкие родственные группы с дефицитом фактора XIII с гемартрозами и в/че-репными кровоизлияниями. Результаты обычных скрининговых тестов на гемостаз у пациентов с дефицитом фактора XIII находятся в норме.
Скрининговые тесты на дефицит фактора XIII основаны на наблюдении, что из-за нарушения перекрестного сшивания растворимость сгустка крови повышена. Нормальный сгусток крови не растворяется в присутствии 5М мочевины, тогда как у пациента с дефицитом фактора XIII сгусток растворяется. Более специфические тесты на определение фактора XIII являются иммунологическими. Период полувыведения фактора XIII составляет 5-7 дней, а уровень гемостатической активности — 2-3%. Для лечения или профилактики эпизодов кровотечения применяется термообработанный лиофилизированный концентрат фактора свертывания XIII.
л) Дефициты антиплазмина и ингибитора активатора плазминогена. Дефицит антиплазмина и ингибитора активатора плазминогена, оба из которых являются антифибринолитическими белками, приводит к увеличению производства плазмина и преждевременному лизису фибриновых сгустков. У пораженных пациентов наблюдается нарушение свертывания крови легкой степени, характеризующееся кожно-слизистыми кровотечениями, но кровоизлияния в суставы встречаются редко. Поскольку результаты обычных гемостатических тестов находятся в норме, дальнейшее обследование пациента с «+» анамнезом кровотечения должно включать время лизиса эуглобулинового сгустка (если возможно), которое измеряет фибринолитическую активность и показывает сокращенные значения при наличии этих дефицитов.
Существуют специфические тесты на α2-антиплазмин и ингибитор активатора плазминогена. Эпизоды кровотечения лечатся с помощью свежезамороженной плазмы; кровотечения в полости рта могут отвечать на антифибринолитическую терапию.
Видео схема активации свертывания крови по внутреннему и внешнему пути - профессор А.Б. Добровольский