Синдром Альпорта — генетически гетерогенное заболевание, в основе которого лежат мутации генов коллагена IV типа — основного компонента базальной мембраны. Соответственно, клинические проявления синдрома Альпорта связаны с нарушениями в строении и функционировании базальной мембраны. Основной симптом — стойкая микрогематурия.
У мальчиков и мужчин неизбежно развиваются протеинурия, артериальная гипертония и почечная недостаточность, тогда как у девочек и женщин почечная недостаточность возникает редко. Поражение почек нередко сочетается с нейросенсорной глухотой и поражением глаз, в частности дегенерацией пигментного эпителия в области желтого пятна сетчатки и лентиконусом.
Примерно в 80% случаев синдром Альпорта наследуется Х-сцепленно и вызван мутацией гена COL4A5, кодирующего а5-цепь коллагена IV типа. Вариант Х-сцепленного синдрома Альпорта с диффузным лейомиоматозом вызван делецией, затрагивающей лежащие рядом гены COL4A5 и COL4A6. У оставшейся части больных заболевание наследуется аутосомно-рецессивно и связано с мутациями генов COL4A3 или COL4A4, расположенных на 2-й хромосоме и кодирующих а3- и а4-цепи коллагена IV типа соответственно.
Аутосомно-доминантный тип синдрома Альпорта встречается редко, он также связан с мутациями генов COL4A3 и COL4A4.
Существует шесть изомеров а-цепи коллагена IV типа, их обозначают от а 1 до а6. Цепи а.1 и а2 присутствуют во всех базальных мембранах, тогда как экспрессия остальных цепей в разных тканях различна. Цепи с аЗ по а5 имеются в тех органах, которые поражаются при синдроме Альпорта: почках, глазах и ушах. В почках а3-, а4- и а5-цепи присутствуют в базальных мембранах клубочков, дистальных канальцев и собирательных трубочек, а также в капсуле клубочка.
У большинства мальчиков с Х-сцепленным синдромом Альпорта в почечных базальных мембранах полностью отсутствуют цепи с а3 по а6. У девочек с этим заболеванием из-за случайного характера инактивации Х-хромосомы экспрессия этих цепей частично сохранена. При аутосомно-рецессивном синдроме Альпорта аЗ- и а4-цепи отсутствуют во всех базальных мембранах, а5-цепь отсутствует только в базальной мембране клубочка, а в капсуле клубочка и базальных мембранах дистальных канальцев, собирательных трубочек и эпидермиса экспрессия а5- и а6-цепей сохранена. В глазах цепи с а3 по а5 имеются в базальных мембранах капсулы хрусталика, роговицы, задней пограничной пластинки, базальной пластинки (мембраны Бруха) и внутренней пограничной мембраны сетчатки. Они присутствуют также во внутреннем ухе: основной мембране внутреннего уха и базальных мембранах спирального выступа, спирального гребешка, внутренней и наружной спиральных борозд.
Сегодня основной метод диагностики синдрома Альпорта — биопсия почки, хотя, возможно, в будущем ее заменят молекулярно-генетические методы. При электронной микроскопии выявляют расслаивание и утолщение базальной мембраны клубочка. Дополнительное исследование аЗ-, а4- и а5-цепей в ткани почки может оказаться полезным в тех случаях, когда данные биопсии сомнительны. Отсутствие а5-цепи в базальной мембране эпидермиса указывает на Х-сцепленный синдром Альпорта, в этом случае биопсия почки необязательна.
Лечения не существует. Х-сцепленный синдром Альпорта встречается у собак, он служит моделью для разработки генотерапии и лекарственных средств. Трансплантация почки, как правило, дает хороший результат. Из-за того что вместе со здоровой почкой больные получают и новые антигены, примерно у 5% больных развивается антительный гломерулонефрит пересаженной почки, который почти всегда приводит к гибели трансплантата.
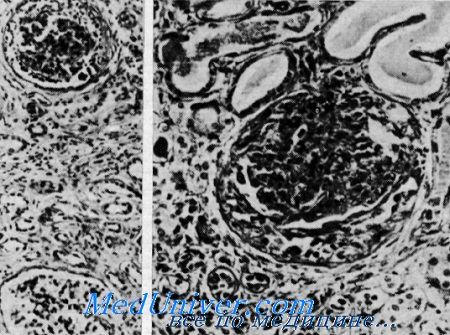
Нефрит и нейросенсорная глухота (синдром Альпорта). Сверху клубочек с утолщенной основной мембраной и слипшейся капсулой в противоположность почти нормальному клубочку внизу. Видны редкие клубочки, имеющие полулупиую форму, расширенные канальцы, содержащие белковые цилиндры. Утолщенная основная мембрана, окружающая атрофичные канальцы в области фиброза (из H. I. Krickstein et al.).