Врожденный нефротический синдром финского типа - клиника, диагностика
Врожденный нефротический синдром финского типа - заболевание наследуется по аутосомно-рецессивному типу и является основной причиной высокой протеинурии у детей первого месяца жизни. Хотя наибольшая распространенность этого заболевания отмечается в Финляндии (1,2 случая на 10 000 беременностей), описано много случаев заболевания у детей других национальностей.
При этом заболевании протеинурия возникает еще внутриутробно, что проявляется повышенным уровнем а-фетопротеина в околоплодных водах. Уже на первой неделе жизни часто возникают отеки. Истощение, тяжелые инфекции и тромбозы обусловливают тяжесть заболевания и высокую смертность, ранее больные погибали на первом году жизни. Сегодня при интенсивном лечении больные могут дожить до того момента, когда им можно провести трансплантацию почки. Выживаемость как трансплантата, так и больных очень высокая.
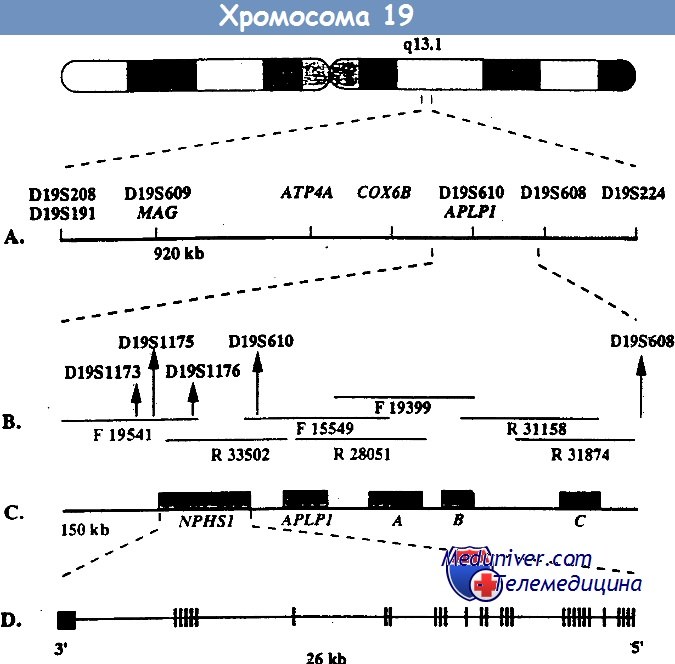
Локус, мутация в котором обусловливает данное заболевание, был найден с помощью позиционного клонирования на длинном плече 19-й хромосомы (19q13.1) и в финских, и в других семьях. При определении нуклеотидной последовательности этого локуса был найден ранее неизвестный ген NPHS1, который избирательно экспрессируется в подоцитах. Продукт этого гена получил название нефрин.
Он относится к молекулам адгезии из суперсемейства иммуноглобулинов. Нефрин локализован в области щелевых диафрагм — видоизмененных плотных контактов между отростками ножек подоцитов. У больных с мутацией гена NPHS1 нет отростков ножек подоцитов и щелевых диафрагм. Это позволяет думать, что именно нефрин является важнейшим компонентом щелевых диафрагм, предотвращающих выход белка из сосудов клубочка.
Среди всех мутаций гена NPHS1 у финнов преобладают две: Fin-major и Fin-minor. Они присутствуют более чем у 90% больных. Мутация Fin-major вызвана делецией двух пар нуклеотидов во 2-м экзоне, который кодирует терминирующий кодон, она встречается примерно у 80% больных финнов.
Мутация Fin-minor — нонсенс-мутация в 26-м экзоне, она встречается примерно у 17% больных финнов. У больных других национальностей встречаются различные мутации по типу делеций, вставок, нонсенс- и миссенс-мутаций, а также мутации, нарушающие сплайсинг. Врожденный нефротический синдром финского типа — основная, но не единственная причина нефротического синдрома на первом месяце жизни.